Defect in Glucose Homeostasis
In uncontrolled diabetes, hyperglycemia is caused by increased hepatic and renal glucose production and decreased glucose utilization in muscle and adipose tissue. Decreased glucose utilization, once considered the major contributor to hyperglycemia in uncontrolled diabetes, is currently believed to play a smaller role than excessive glucose production.
Figure 74.2 depicts the hepatic and renal contribution to endogenous glucose production in conscious normal and diabetic dogs.
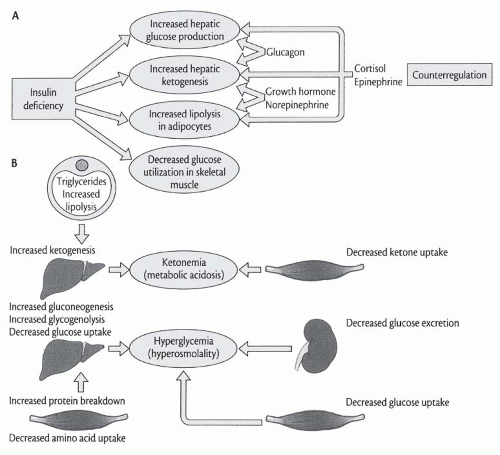
13 The rise in the glucagon/insulin ratio in plasma characteristic of uncontrolled diabetes activates key enzymes that accelerate the rates of both glycogenolysis and gluconeogenesis. The increased ratio also promotes glucose overproduction by modulating the effects of other hormones, availability of substrate, and rates of fatty acid oxidation (
Fig. 74.1). Volume depletion secondary to hyperglycemia-induced osmotic diuresis reduces the urinary loss of glucose, thereby worsening hyperglycemia.
An important contributor to the development of hyperglycemia in uncontrolled diabetes may be the prevailing acidemia.
14,
15,
16,
17,
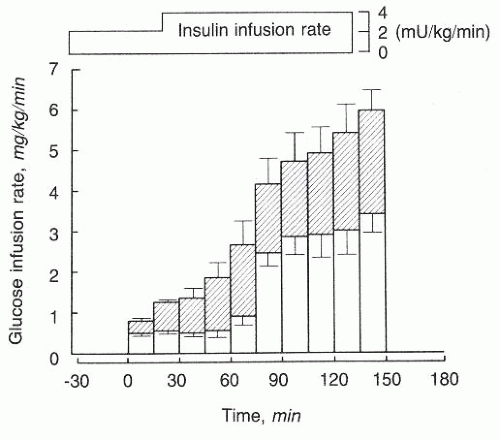
18 In animals with hypercapnia-induced acidemia, for example, a substantially smaller glucose infusion rate maintains euglycemia as compared to dogs without respiratory acidosis during constant insulin infusion, reflecting less glucose entry into cells for a given insulin level (
Fig. 74.3).
19 Although the sympathetic surge characteristic of acidemia undoubtedly contributes to glucose intolerance, adrenergic blockade during acute respiratory acidosis does not prevent the disturbed glucoregulation. Nor do plasma levels of insulin fall during acute respiratory acidosis. In fact, acidemia reduces tissue extraction of insulin and, more specifically,
insulin uptake by the liver.
19 Although plasma glucagon levels also increase during metabolic or respiratory acidosis, the glucagon/insulin ratio in the portal circulation remains unchanged, thereby reducing the possible role of glucagon in the hyperglycemia of acidemic states. The weight of the evidence suggests that the hyperglycemia of acidemia is mediated by reduction of insulin binding to its receptor and decreased tissue sensitivity to the hormone.
19,
20,
21,
22The defect in glucose homeostasis observed in uncontrolled diabetes might lead to either hyponatremia or hypernatremia.
23,
24,
25 The hyperglycemia-induced increase in effective osmotic pressure of the extracellular fluid (ECF) triggers a shift of water out of cells, most prominently skeletal muscle, which reduces serum [Na
+]. An increase of 100 mg per dL (5.6 mmol per L) in the glucose concentration decreases serum [Na
+] by approximately 1.7 mEq per L, the end result being a rise in serum osmolality by approximately 2.0 mOsm per kg H
2 O.
25,
26 The resulting ECF expansion is, however, brief due to simultaneous renal and extrarenal loss of fluids. The hyperglycemia-induced increase in the filtered load of glucose exceeds the renal tubular reabsorptive capacity resulting in substantial glucosuria—one of the hallmarks of DKA. In turn, glucosuria causes osmotic diuresis that results in urinary losses of 75 to 150 mL per kg of water and 7 to 10 mEq per kg of Na
+ and Cl˜ over an entire episode of DKA.
Because the total of the Na
+ and K
+ concentrations in the urine falls short of that in serum, osmotic diuresis elevates serum [Na
+] and [Cl
–]; moderation of hyponatremia or frank hypernatremia can ensue.
23,
27 However, other factors also act to modify the serum [Na
+] and [Cl
–].
27 Some Na
+ enters cells replacing cellular K
+ losses, thereby decreasing serum [Na
+]. Urinary losses of Na
+ as ketone salts tend to increase serum [Cl
–], whereas selective Cl
– depletion during vomiting tends to cause hypochloremia. Further, the intake of fluid and electrolytes (sodium, potassium, and chloride) influences serum [Na
+] and [Cl
–]. Differences in the magnitude of these phenomena from one patient to another account for the variability in serum electrolyte composition observed at presentation.
Table 74.2 reviews the admitting laboratory values of patients with DKA.
8 Note that serum [Na
+] is usually depressed; the rare presence of hypernatremia is indicative of a profound water depletion, usually seen in the most critically ill patients.
Prerenal azotemia due to volume depletion is almost always present in uncontrolled diabetes and is usually reversible, but occasionally it can progress to acute tubular necrosis.
28,
29 The levels of plasma urea nitrogen, creatinine, total protein, uric acid, hematocrit, and hemoglobin can all be elevated on admission for DKA, a reflection of ECF contraction, and/or renal dysfunction, but they normalize swiftly after volume repletion.
Defect in Water Homeostasis
Examination of the defect in water homeostasis that accompanies diabetes mellitus requires a brief overview of this topic.
23,
24,
25 The disorders of salt and water balance may be classified into three major categories: (1) abnormalities in the size of body fluid compartments, (2) disturbances in the tonicity of body fluids, and (3) a selective deficit or excess of chloride with respect to sodium.
25 The first group of disorders comprises an enlargement (“volume expansion”) and a reduction (“volume depletion or contraction”) in the size of the ECF compartment, which are produced by a combined salt and water excess and a combined salt and water deficit, respectively. Disturbances in the tonicity of body fluids include increases (e.g., hypernatremia) and decreases (e.g., hypotonic hyponatremia) in the effective osmolality of body fluids. In contrast to the first group in which salt and water excess or deficit develops with normal proportionality, a discordant abnormality in salt and water balance occurs in disorders of body fluid tonicity. The third group of salt and
water disorders is characterized by an abnormal relationship between the [Na
+]
p and [Cl˜]
p (plasma concentration). Although the [Na
+]
p is generally maintained within normal limits in these disorders because it is dependent on overall water homeostasis, the [Cl˜]
p is either abnormally low or high. The major representatives of a selective deficit or excess of chloride with respect to sodium are hypochloremic metabolic alkalosis and hyperchloremic metabolic acidosis, respectively. These acid-base disorders are reviewed in the section dealing with such abnormalities.
Disturbances in salt balance are the primary causes of volume excess and depletion, whereas disorders in water balance are responsible for the development of the tonicity disorders, hypertonicity (hypernatremia), and hypotonicity (hyponatremia). Because sodium chloride (NaCl) excess only transiently increases tonicity, leading to augmented antidiuretic hormone (ADH) (i.e., arginine vasopressin) secretion and secondary water retention, hypernatremia is not clinically observed. Expansion of the ECF volume, instead, is the hallmark of a primary NaCl excess. In a comparable fashion, NaCl deficit only transiently decreases tonicity, inhibiting ADH secretion with secondary increase in water excretion, so that hyponatremia is not observed, whereas volume depletion becomes the major manifestation of this electrolyte imbalance. A primary and exclusive disturbance in water balance, deficit and excess, causes hypertonicity (hypernatremia) and hypotonicity (hyponatremia), respectively, but does not produce a major alteration in the size of the fluid compartments because the latter is primarily determined by the osmolar content in each compartment, and the change in water content is distributed throughout the body fluids.
25Dysnatremias in Diabetes Mellitus. A defect in water homeostasis in patients with diabetes mellitus might lead to either hypotonic hyponatremia or hypernatremia in response to positive or negative water balance, respectively.
25 Water and electrolyte losses caused by vomiting or diarrhea are commonly encountered in uncontrolled diabetes as well as patients with diabetes experiencing target organ damage in the alimentary tract (e.g., gastroparesis, nocturnal diarrhea). In addition, excessive urinary fluid losses may develop as a result of osmotic diuresis, use of diuretics, adrenal insufficiency, or other causes. Whether hypotonic hyponatremia or hypernatremia develops is dependent on the concomitant water intake. Hypernatremia might be observed if water intake is insufficient, whereas a large salt-free fluid intake might lead to hyponatremia. Long-standing diabetes mellitus commonly predisposes or leads to heart failure, renal failure, or both, thereby impairing renal water excretion that may lead to hypotonic hyponatremia. Concomitant medication, including diuretics, might also play a role in the development of hyponatremia.
Abnormal [Na+]p can produce signs and symptoms owing to central nervous system dysfunction and the clinical manifestations elicited by opposite changes in tonicity are remarkably similar, except for seizures that are mostly caused by cerebral edema secondary to dilutional (hypotonic) hyponatremia. When the patient’s osmoregulating mechanisms (thirst, changes in water intake and ADH levels, renal water retention or excretion) fail, an increase or decrease in plasma tonicity and [Na+]p develops. Plasma hypertonicity induces brain water loss, whereas hypotonicity produces water gain in this organ, accompanied in both cases by parallel volume changes. As a defense mechanism to correct brain volume changes, the intracellular osmolytes of this organ increase in hypernatremia and decrease in hypotonic hyponatremia. The adaptive increase in brain osmolytes reflects a modest increase in cellular K+ and accumulation of organic solutes (e.g., glutamine, glutamate, and other organic metabolites), which are referred to as idiogenic osmoles. Conversely, the adaptive decrease in brain osmolytes reflects a decrease in cellular K+ accompanied by a diminished concentration of idiogenic osmoles. These secondary responses of the brain to altered extracellular tonicity can be demonstrated within a few hours of the initiation of abnormal tonicity and are complete within a few days.
Hypotonic or Dilutional Hyponatremia. It represents an excess of water in relation to existing sodium stores, which can be decreased, essentially normal, or increased. Retention of water most commonly reflects the presence of conditions that impair renal excretion of water; in a minority of cases, however, it is caused by excessive water intake, with a normal or nearly normal excretory capacity.
Conditions of impaired renal excretion of water are categorized according to the characteristics of the ECF volume, as determined by clinical assessment. Decreased ECF volume can result from renal sodium loss (e.g., glucosuriainduced osmotic diuresis) or extrarenal sodium loss (e.g., vomiting). Conditions with essentially normal ECF volume include thiazide diuretics, syndrome of inappropriate secretion of antidiuretic hormone, decreased intake of solutes, hypothyroidism, and glucocorticoid insufficiency. Increased ECF volume with hyponatremia can be observed in pregnancy, renal failure, congestive heart failure, cirrhosis, and nephrotic syndrome. With the exception of renal failure, these conditions are characterized by high plasma concentrations of ADH despite the presence of hypotonicity; arterial underfilling induces baroreceptor-mediated nonosmotic release of ADH that overrides the osmotic regulation of the hormone, thereby impairing urinary dilution and causing hyponatremia. Depletion of potassium accompanies many of these disorders and contributes to hyponatremia because the sodium concentration is determined by the ratio of the “exchangeable” (i.e., osmotically active) portions of the body’s sodium and potassium content to total body water. Patients with hyponatremia induced by thiazides can present with variable hypovolemia or apparent euvolemia, depending on the magnitude of the sodium loss and water retention.
Excessive water intake can cause hyponatremia by overwhelming normal water excretory capacity (e.g., 15 to 20 L per day). Frequently, however, psychiatric patients with excessive water intake have plasma arginine vasopressin concentrations that are not fully suppressed and urine that is not maximally dilute, thus contributing to water retention.
The optimal treatment of hypotonic hyponatremia requires balancing the risks of hypotonicity against those of therapy.
23 The presence of symptoms and their severity largely determine the pace of correction. Patients with symptomatic hyponatremia and dilute urine (osmolality, <200 mOsm per kg water) but with less serious symptoms usually require only water restriction and close observation. Severe symptoms (e.g., seizures or coma) call for infusion of hypertonic saline. On the other hand, patients who have symptomatic hyponatremia with concentrated urine (osmolality >200 mOsm per kg water) in association with a hypovolemic state are best treated with isotonic saline; those having clinical euvolemia or hypervolemia require infusion of hypertonic saline.
There is no consensus about the optimal treatment of symptomatic hyponatremia.
23 Nevertheless, correction should be of a sufficient pace and magnitude to reverse the manifestations of hypotonicity but not be so rapid and large as to pose a risk of the development of central pontine myelinolysis. Osmotic demyelination is a serious disorder and can develop one to several days after aggressive treatment of hyponatremia by any method, including water restriction alone. Shrinkage of the brain triggers demyelination of pontine and extrapontine neurons that can cause neurologic dysfunction, including quadriplegia, pseudobulbar palsy, seizures, coma, and even death. Hepatic failure, potassium depletion, and malnutrition increase the risk of the complication. Physiologic considerations indicate that a relatively small increase in the serum [Na
+], on the order of 5%, should substantially reduce cerebral edema in patients with symptomatic hypotonic hyponatremia. Even seizures induced by hyponatremia can be stopped by rapid increases in the serum [Na
+] that average only 3 to 7 mEq per L. Most reported cases of osmotic demyelination occurred after rates of correction that exceeded 12 mEq per L per day were used, but isolated cases occurred after corrections of only 9 to 10 mEq per L in 24 hours or 19 mEq per L in 48 hours. After weighing the available evidence and the all-too-real risk of overshooting the mark, we recommend a targeted rate of correction that does not exceed 8 mEq per L on any day of treatment. Remaining within this target, the initial rate of correction can still be 1 to 2 mEq per L per hour for several hours in patients with severe symptoms.
The rate of infusion of the selected solution can be derived expediently by applying the following equations:
Equation 74.1 projects the impact of 1 L of any infusate on the patient’s [Na
+]
s
Equation 74.2 is a simple derivative of
equation 74.1 and projects the impact of 1 L of any infusate containing sodium and potassium on the patient’s [Na
+]
s
The preceding equations project the change in serum [Na
+] elicited by the retention of 1 L of any infusate.
30 Dividing the change in serum sodium targeted for a given treatment period by the output of this equation determines the volume of infusate required, and hence the rate of infusion. Although water restriction ameliorates all forms of hyponatremia, as explained, it is not the optimal therapy in all cases.
Corrective measures for nonhypotonic hyponatremia are directed at the underlying disorder rather than at the hyponatremia itself. Administration of insulin is the basis of treatment for uncontrolled diabetes, but deficits of water, sodium, and potassium also should be corrected.
Hypernatremia. Defined as a rise in the [Na
+]
p to a value exceeding 145 mEq per L, it represents a deficit of water in relation to the body’s sodium stores, which can result from a net water loss or a hypertonic sodium gain.
24 Net water loss accounts for the majority of cases of hypernatremia. It can occur in the absence of a sodium deficit (pure water loss) or in its presence (hypotonic fluid loss). Net water loss can result from pure water (e.g., hypodipsia, diabetes insipidus) or hypotonic fluid loss, the latter secondary to renal, gastrointestinal, or cutaneous causes.
An equation
31 that allows projection of the expected Δ[Na
+]
s in response to losing 1 L of fluid (fl) of variable electrolyte content from the renal or extrarenal route is as follows:
Multiplying the output of the equation by the volume of the fluid loss in liters provides a quantitative estimate of the impact of the fluid loss on [Na+]s. Obviously, application of this equation has greater practical value in the presence of large fluid losses (e.g., large gastrointestinal drainage, polyuria).
Hypertonic sodium gain usually results from clinical interventions (e.g., sodium bicarbonate infusion, hypertonic enemas) or accidental sodium loading. Signs and symptoms of hypernatremia largely reflect central nervous system dysfunction and are prominent when the increase in the serum [Na
+] is large or occurs rapidly (i.e., over a period of hours). Most outpatients with hypernatremia are either very young or very old. Common symptoms in infants include hyperpnea, muscle weakness, restlessness, a characteristic highpitched cry, insomnia, lethargy, and even coma. Convulsions are typically absent except in cases of inadvertent sodium loading or aggressive rehydration. Brain shrinkage induced by hypernatremia can cause vascular rupture, with cerebral
bleeding, subarachnoid hemorrhage, and permanent neurologic damage or death. Brain shrinkage is countered by an adaptive response that is initiated promptly and consists of solute gain by the brain that tends to restore lost water.
Proper treatment of hypernatremia requires a twopronged approach: addressing the underlying cause and correcting the prevailing hypertonicity.
24 Managing the underlying cause may mean stopping gastrointestinal fluid losses; controlling pyrexia, hyperglycemia, and glucosuria; withholding lactulose and diuretics; treating hypercalcemia and hypokalemia; moderating lithium-induced polyuria; or correcting the feeding preparation. In patients with hypernatremia that has developed over a period of hours (e.g., those with accidental sodium loading) rapid correction improves the prognosis without increasing the risk of cerebral edema, because accumulated electrolytes are rapidly extruded from brain cells. In such patients reducing the [Na
+]
p by 1 mEq/L/hour is appropriate. A slower pace of correction is prudent in patients with hypernatremia of longer or unknown duration, because the full dissipation of accumulated brain solutes occurs over a period of several days. In such patients, reducing the [Na
+]
p at a maximal rate of 0.5 mEq/L/hour prevents cerebral edema and convulsions. Consequently, we recommend a targeted fall in the [Na
+]
p of 10 mEq/L/day for all patients with hypernatremia except those in whom the disorder has developed over a period of hours. The goal of treatment is to reduce the [Na
+]
p to 145 mEq per L. Because ongoing losses of hypotonic fluids, whether obligatory or incidental, aggravate the hypernatremia, allowance for these losses must also be made.
The preferred route for administering fluids is the oral route or a feeding tube; if neither is feasible, fluids should be given intravenously. Only hypotonic fluids are appropriate, including pure water, 5% dextrose, 0.2% NaCl (referred to as one quarter isotonic saline), and 0.45% NaCl (one half isotonic saline). The more hypotonic the infusate, the lower the infusion rate required. The volume should be restricted to that required to correct hypertonicity because the risk of cerebral edema increases with the volume of the infusate. Except in cases of frank circulatory compromise, 0.9% NaCl (isotonic saline) is unsuitable for managing hypernatremia.
After selecting the appropriate infusate, the physician must determine the rate of infusion. This can be easily calculated with the use of equations 74.1 and 74.2 which estimate the change in the serum sodium concentration caused by the retention of 1 L of any infusate. The sole indication for administering isotonic saline to a patient with hypernatremia is a depletion of ECF volume sufficient to cause substantial hemodynamic compromise. Even in this case, after a limited amount of isotonic saline has been administered to stabilize the patient’s circulatory status, a hypotonic fluid (i.e., 0.2% or 0.45% NaCl) should be substituted in order to restore normal hemodynamic values while correcting the hypernatremia. If a hypotonic fluid is not substituted for isotonic saline, the ECF volume may become seriously overloaded.
Defect in Sodium Homeostasis
The quantity of solutes in each of the main fluid compartments determines its size, so that deficit or excess of solutes in a particular space will shrink or swell that space in comparison with the other compartments.
25 The partition of water is determined by the osmotic activity of the solutes confined to each body compartment. One major solute is responsible for the size of each fluid compartment. These solutes are potassium, sodium, and proteins, for the intracellular, extracellular, and intravascular spaces, respectively. Because the hydraulic permeability of most cell membranes is very high, solute-free water freely and rapidly moves across all body compartments.
Body stores of NaCl are determined by the balance of its intake and excretion. Under normal circumstances, NaCl intake is derived from the diet and its excretion occurs by urinary loss. A positive NaCl balance (intake exceeds excretion) increases salt stores, whereas a negative one (excretion exceeds intake) decreases salt stores. The effect of increased NaCl stores is expansion of ECF volume, whereas decreased NaCl stores lead to a reduced ECF volume. Thus, an NaCl deficit in body fluids (e.g., vomiting, diarrhea) reduces ECF volume, including the intravascular compartment. By contrast, NaCl excess (e.g., congestive heart failure) expands ECF volume and can produce overt peripheral edema and accumulation of fluid in major body cavities (pleural effusion, ascites). A major decrease in serum protein concentration (mostly albumin) diminishes intravascular volume and promotes expansion of the interstitial compartment (e.g., nephrotic syndrome, hepatic cirrhosis). Diabetes mellitus is a common cause of both volume depletion and volume expansion. The former disturbance is characteristically observed in the course of severe metabolic complications of this disease, namely, DKA and NKH. Conversely, volume expansion is observed in patients having chronic diabetic complications, including congestive heart failure, nephrotic syndrome, and renal failure.
Volume Depletion. Volume depletion in diabetic patients can result from fluid loss (e.g., renal and/or extrarenal) or from fluid sequestered into a “third space” (e.g., acute pancreatitis). Renal losses may occur in the presence of normal intrinsic renal function (e.g., osmotic diuresis caused by glucosuria or urea diuresis, adrenal insufficiency, diuretics) or in acute and chronic renal disease (e.g., acute tubular necrosis, diabetic glomerulosclerosis). Osmotic diuresis owing to renal excretion of glucose can produce a large natriuresis, leading to volume depletion. Patients with significant hyperglycemia, including those with DKA or nonketotic coma, may have a fluid deficit of 10% or more of body weight. Extrarenal losses include those from the gastrointestinal tract (e.g., vomiting, diarrhea, gastrointestinal suction, fistulas) and those from the skin (sweat, burns, extensive
skin lesions). Fluid sequestration into a third space occurs with abdominal accumulation (e.g., intestinal obstruction, pancreatitis, peritonitis), bleeding, skeletal fractures, and obstruction of a major venous system.
The patient’s history, physical examination, and laboratory data are critical elements in the evaluation of volume depletion, allowing the physician to (1) assess the severity of the deficit, and (2) establish its cause. Immediate recognition of hypovolemic shock is of utmost importance, because rapid intravascular volume expansion might prevent tissue injury and death. Evaluation of its severity allows establishment of the rate of infusion and the total fluid requirements. Recognition of the factors responsible for fluid loss permits initiation of specific therapeutic measures to correct the volume depletion.
25Patients with volume depletion have signs and symptoms related to (1) the process responsible for volume depletion, and (2) the hemodynamic consequences of fluid loss. Through the first group of manifestations it is possible to recognize the cause of volume depletion, such as loss or sequestration of fluid. The second group of signs and symptoms includes hypotension, decreased cardiac output, and tachycardia owing to intravascular volume depletion. In addition, diminished tissue perfusion produces altered mental status, generalized weakness, and occasionally severe organ damage (e.g., acute tubular necrosis, cerebral ischemia, myocardial infarction).
The severity of volume deficit may be estimated through evaluation of blood pressure, heart rate, neck veins and venous pressure, skin turgor, moistness of mucous membranes, changes in body weight, and blood and urine indices. If volume depletion results from mechanisms other than hemorrhage, the fluid loss produces hemoconcentration with increased hematocrit (Hct). The ECF volume deficit can be estimated in states of a primary extravascular fluid loss from the rise in Hct as follows:
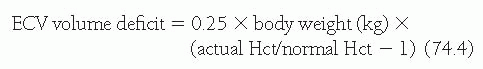
where 0.25 represents the fraction of ECF per kg of body weight (250 mL per kg). Because the normal range of Hct is relatively wide (38% to 45%), the patient’s baseline Hct usually is unknown, and blood loss may have occurred, the reliability of changes in Hct is only modest. Therefore, a precise estimation of volume deficit is difficult. The loss of body weight from its baseline level (body weight prior to the episode of volume depletion) is a clinically useful index to estimate volume deficit, as follows:
The change in body weight is unreliable for the estimation of fluid deficit in patients with “third space” sequestration. If [Na+]p remains within normal limits, the weight loss in kilogram truly represents loss of isotonic fluid. Volume deficit accompanied by hypernatremia or hyponatremia indicates the existence of a disproportionate water loss compared to Na+ loss.
Pertinent blood indices that are most useful in the diagnosis and management of volume depletion include: (1) blood urea nitrogen (BUN) and serum creatinine levels; (2) Hct, total plasma protein, and/or albumin values; and (3) levels of serum electrolytes, including Na+, K+, Cl˜, and total carbon dioxide (almost identical to plasma [HCO3˜]). In volume depletion, BUN and plasma creatinine increase because of an overall depression of renal function, manifested by oliguria, and reduced glomerular filtration rate (GFR) and renal plasma flow. Increased plasma creatinine is caused by a reduced GFR (when muscle necrosis, which could release this substance into the circulation, is absent). Conversely, an elevated BUN, not accompanied by increased plasma creatinine and reduced GFR, reflects enhanced renal reabsorption of urea accompanied by increased salt and water reabsorption. Consequently, the ratio of BUN over plasma creatinine increases from its normal value of 10:1 to 15:1 or more. Hematocrit and concentration of plasma proteins also can increase in volume depletion, a process referred to as hemoconcentration. Alterations in serum electrolytes are commonly observed and they depend on the composition of the fluid lost (e.g., vomiting produces hypokalemia and metabolic alkalosis) as well as the concomitant water and electrolyte intake.
With respect to fluid therapy in volume depletion, considering that oral intake is the physiologic pathway for the entry of fluids, this route should be always considered. Oral replacement therapy is effective, relatively inexpensive, and noninvasive; does not require hospitalization; and saves several million patients (mostly children in developing nations) each year from death. Nevertheless, the presence of vomiting, ileus, or altered mental status precludes its use, mandating intravenous administration of fluid. Most frequently, however, volume repletion in hospitalized patients is performed by the parenteral (intravenous) route.
Volume repletion should be promptly secured because severe volume depletion frequently produces a major reduction in intravascular volume and hypovolemic shock. The type of fluid to be used depends on the cause of volume depletion. Hypovolemia caused by bleeding (e.g., peptic ulcer, rupture of aortic aneurysm) must be treated with blood products or plasma volume expanders (e.g., packed red cells, albumin, or dextran solutions), whereas that resulting from renal or extrarenal losses and fluid sequestration in body cavities (e.g., ileus, ascites) must be treated with saline, dextrose in saline, or Ringer’s solution. Plasma volume expanders can be used in the initial phase of treatment to secure a more rapid restoration of hemodynamic status in all patients with shock.
Various intravenous solutions can be selected in fluid therapy.
25 The most commonly used intravenous fluids consist of a NaCl-containing solution (NaCl 0.23%, 0.45%, and 0.9%, known as 1/4 normal saline, 1/2 normal saline, and
normal saline, respectively) with or without 5% dextrose. The term normal used in reference to intravenous solutions does not imply “normality” (chemical notation) but simply refers to the isotonicity of intravenous solutions with respect to body fluids. It is more proper to refer to these solutions as 1/4 isotonic saline, 1/4 isotonic saline, and isotonic saline. Although 5% dextrose in water is isotonic with body fluids, the glucose is metabolized so that this solution provides solute-free water without effective long-lasting osmoles (yet providing some caloric intake). The NaCl added to intravenous solutions provides effective osmoles that are preferentially retained in ECF. The efficacy of the various solutions with respect to volume deficit correction is a function of their NaCl concentration, with normal saline as the most effective one and dextrose in water without NaCl the least effective. The selection of intravenous solution is also determined by the patient’s [Na
+]
p ; hypernatremic patients are most frequently treated with NaCl-free solutions (e.g., 5% dextrose in water), whereas those with hyponatremia are usually given isotonic saline or hypertonic (e.g., NaCl 3.0%) saline solutions. It is important to realize the expected changes in the volume of ECF and ICF in response to various solutions. The infusion of normal saline expands the ECF exclusively (ECF volume increment is identical to the volume infused); thus, ICF volume remains unaltered. The infusion of 1/2 isotonic saline expands both the ECF and ICF, with the former receiving 73% and the latter 27% of the volume load. Finally, a salt-free water infusion (e.g., 5% dextrose in water) will also expand both the ECF and ICF, but in this case, the latter receives 60% of the volume load. In summary, a pure water infusion expands all body compartments but predominantly the ICF, whereas isotonic saline expands the ECF exclusively.
23Because patients in hypovolemic shock are at immediate risk of death or ischemic tissue injury, the initial fluid infusion should be at the maximal flow allowed by the intravenous catheter (“wide open”); once blood pressure and tissue perfusion return to acceptable levels, the rate must be diminished to approximately 100 mL per hour to minimize the risk of pulmonary edema, owing to rapid intravascular expansion. Patients with acceptable hemodynamic parameters should receive fluid at initial rates of 100 to 200 mL per hour, with subsequent reduction after 6 to 12 hours to rates of about 100 mL per hour, to secure gradual repletion of all fluid compartments without imposing undue stress on the circulation. Exceptions to these rules are patients with extreme volume depletion (e.g., DKA, NKH) or large ongoing fluid losses (e.g., continuous drainage of large volume of gastrointestinal secretions, postobstructive diuresis, and diabetes insipidus) who might require fluids at a higher rate of infusion as described in the corresponding section of this chapter.
Proper monitoring of fluid replacement therapy is accomplished by evaluation of arterial blood pressure, presence of collapsed or distended neck veins, and urine output to establish the optimum rate of fluid replacement. Additional information might be necessary in critically ill patients, including monitoring of left- and right-sided heart filling pressures, blood pressure measurement through an intra-arterial line, arterial and/or venous blood gas analysis, and sequential chest X-ray films to detect pulmonary venous congestion and interstitial edema.
Volume Expansion. A syndrome of volume expansion caused by overt salt and water retention is commonly observed in long-standing diabetes mellitus.
32,
33 Both forms of generalized edema, the so-called primary as well as the secondary types, are encountered. In primary edema, renal retention of salt and water is the initial event that leads to expansion of ECF volume (e.g., diabetic glomerulosclerosis with reduced GFR and avid tubular reabsorption of salt and water). In secondary edema, also called underfill edema, the presence of renal hypoperfusion, owing to decreased “effective arterial circulating blood volume,” initiates salt and water retention by the kidney (e.g., diabetes mellitus with congestive heart failure). Thus, the kidney is always involved in the development of positive salt and water balance that leads to generalized edema.
25 It must be recognized that salt and water retention, owing to primary renal disease and congestive heart failure, is the main cause of generalized edema and normal or near-normal serum albumin. Absence of proteinuria argues against renal disease as the primary cause of fluid retention. Patients with heart failure usually have either minimal or mild urinary protein excretion (1
+ or 2
+ on dipstick determination), whereas those with nephrotic syndrome have, as a rule, severe proteinuria (4+ dipstick). The fluid retention observed in nephrotic syndrome appears to occur as a combination of primary and secondary edema.
The management of localized and generalized edema must be directed, if possible, at the primary cause of fluid accumulation. Effective treatment of the primary cause leads to resolution of the edema. Therapy of the primary process in congestive heart failure can involve the use of afterloadreducing agents, digoxin, and diuretics. Patients with generalized edema most frequently require treatment of the fluid overload in addition to that directed at the primary disease. Correction of fluid overload involves restriction of dietary NaCl, and if this is unsuccessful, the use of diuretic therapy. In addition, both localized and generalized edema are ameliorated by bed rest and elevation of the edematous body area. The management of generalized edema caused by congestive heart failure, nephrotic syndrome, and diabetic glomerulosclerosis is examined in detail in other chapters.
Defect in Potassium Homeostasis
The levels of total body K
+ stores are established by the external K
+ balance, which in turn is determined by the difference between K
+ intake and excretion. The internal K
+ balance refers to the control mechanisms for the distribution of total body K
+ stores between the ICF and the ECF. The major factors that alter internal K
+ balance include hormones (insulin, catecholamines), the acidity of body fluids, the levels of other electrolytes, the tonicity of body fluids,
and drugs.
34Insulin is a major modulator of extrarenal K
+ homeostasis and promotes K
+ uptake in many cell types, including those from skeletal muscle and liver. The hypokalemic action occurs at very low concentrations of insulin and is independent of the effect of insulin on glucose uptake. The precise mechanism of this action remains to be fully defined but appears to involve the activation of several transport proteins, including stimulation of the Na
+-K
+-ATPase, stimulation of the Na
+-H
+ exchanger, and changes in ionic conductance of certain K
+ channels.
35 Direct stimulation of the Na
+-K
+ pump by insulin induces the translocation of K
+ into the cell interior (entry of two K
+ and exit of three Na
+). This action results in hyperpolarization of the membrane potential (a more negative cell interior). Such hyperpolarization of the cell membrane establishes a new electrical gradient, which favors cellular K
+ entry, and deactivates K
+ channels, which inhibits cellular K
+ exit. Thus, the secondary effects of insulin on the membrane potential increase the hypokalemic action of this hormone.
By stimulating the Na+-H+ exchanger, insulin promotes the cellular entry of Na+ and the cellular exit of H+. The entry of Na+ increases the intracellular [Na+], which further stimulates the Na+-K+-ATPase. The cellular exit of H+ results in cytosolic alkalinization, which in turn increases the K+-binding capacity for intracellular anions and stimulates the Na+-K+ pump, therefore favoring cellular K+ loading.
A third mechanism for the hypokalemic effect of insulin is mediated from its action on K+ channels. Insulin controls gating of the inward rectifier K+ channel of skeletal muscle. This channel is responsible for most of the K+ conductance of the skeletal muscle in the resting state. It allows K+ to flow into cells much more easily than it exits from them. Consequently, when the cell membrane is hyperpolarized, the high inward conductance facilitates cellular K+ entry, whereas when the cell membrane is depolarized, the low outward conductance reduces K+ exit from cells. Insulin exaggerates the inward rectifying properties of this class of K+ channel by a dual effect of stimulation of K+ entry and depression of K+ exit.
Glucagon also has significant effects on internal K
+ balance and plasma potassium levels.
36 Glucagon induces glycogen breakdown in the hepatocytes, releasing glucose and K
+; therefore, high glucagon levels can elicit a transient increase in [K
+] p . An increase in plasma glucagon in acute metabolic acidosis has been described and this hormonal response might play a role in acidosis-induced hyperkalemia.
35K
+ Depletion with Hyperkalemia. The development of uncontrolled diabetes, including DKA, is usually accompanied by varying degrees of total body potassium depletion, which results from multiple causes, including massive kaliuresis secondary to glucosuria, decreased intake, and frequent vomiting. However, plasma potassium levels are rarely low at the time of hospitalization, ranging in most instances from normal to high levels and occasionally attaining dangerously elevated values. This paradoxical relationship has been classically attributed to the concomitant changes in blood acidity that would affect a shift of potassium out of the cells in exchange for hydrogen ions moving intracellularly.
37 However, several of the metabolic derangements observed in patients presenting with DKA are known to alter potassium metabolism and may contribute to the development of hyperkalemia. Endogenous ketoacidemia and hyperglycemia correlate with increased plasma potassium concentration on admission in patients with DKA.
36 However, exogenous ketoacidemia and hyperglycemia in the otherwise normal experimental animal fails to increase plasma potassium levels,
38,
39 suggesting that the insulin deficit per se is the major cause of the hyperkalemia that develops in DKA.
36Serum pH and bicarbonate levels are known to alter plasma potassium levels. Whereas some studies indicated that the changes in plasma potassium concentration observed during acute acid-base disorders are consequent to the attendant changes in plasma pH, others showed that a low plasma bicarbonate concentration, under isohydric conditions, may induce hyperkalemia.
40,
41 Increased effective serum osmolality is another abnormality characteristic of DKA that may affect serum potassium; extracellular hypertonicity resulting from the infusion of saline, mannitol, or glucose results in the translocation of potassium-rich cell water to the extracellular compartment.
42 Hyperglycemia of either endogenous or exogenous origin unaccompanied by ketoacidosis results in hyperkalemia in insulindeficient diabetics, especially when hypoaldosteronism also is present.
39As previously described, glucagon may also play a role in the hyperkalemia of DKA. This hormone may cause an increased potassium output from the liver, an effect that is usually transient because of the counterregulatory enhancement of insulin secretion. However, in the presence of an impaired insulin secretion, as in patients with DKA, increments in plasma glucagon levels may result in uncontrolled hyperkalemia.
43An additional mechanism that may be involved in the deranged potassium homeostasis observed in diabetes mellitus is the sympathetic nervous system. Potassium tolerance has been found to be markedly impaired in chemically sympathectomized animals, but is improved in animals given a simultaneous infusion of epinephrine.
44 The effects of the adrenergic agents on the internal potassium balance are mediated by their effect on the plasma levels of insulin and glucagon, and a direct cellular effect on K
+ transport. Therefore, any physiologic condition or pharmacologic maneuver that blocks the β-adrenergic system could result in hyperkalemia, particularly during states of increased potassium load. Diabetic patients may have a suboptimal epinephrine response or altered peripheral sympathetic activity, resulting in potassium movement from the intracellular to
the extracellular space as well as an impairment in cellular entry of potassium.
K+ Depletion with Hypokalemia . Hypokalemia can result from the redistribution or depletion of K
+ stores. The hypokalemia that results from redistribution is caused by cellular uptake of K
+ from the ECF; K
+ redistribution can occur simultaneously with K
+ depletion so that the two processes leading to hypokalemia can have additive effects. The hypokalemia observed with K
+ depletion is characterized by a reduction in the K
+ content of all body fluids.
34
Potassium depletion can occur with diabetes mellitus when dietary K+ intake is very low and therefore fails to counterbalance the obligatory urinary K+ losses associated with glucosuria. However, if K+ losses are abnormally high, potassium depletion might develop in association with a normal dietary K+ intake. Potassium losses may be renal or extrarenal, but a combination of losses is commonly encountered. Total body K+ deficit results in a greater absolute reduction of K+ content in ICF than in ECF. Nevertheless, the percent deviation in K+ content is considerably smaller in ICF than in ECF. In a similar fashion, the decrease in intracellular [K+] with K+ depletion is significantly smaller than the decrease in [K+]p . With respect to the relationship between [K+]p and the degree of K+ deficit, a linear relationship with a slope of 0.3 mEq/L per 100 mEq of K+ deficit [A[K+] p /ΔK+ stores] has been described for patients with K+ depletion in the absence of redistribution of K+ stores. According to this relationship, a K+ depletion of 10% of total body K+ stores (350 mEq) produces a decrease in [K+]p of approximately 1 mEq per L.
Diabetic gastroparesis is a common cause of vomiting leading to fluid and electrolyte losses. Protracted vomiting leads to hypokalemia that is largely caused by increased renal K+ excretion. The increased kaliuresis is owing to HCO3˜ excretion consequent to HCl depletion (metabolic alkalosis) and to secondary hyperaldosteronism resulting from ECF volume depletion. The direct loss of K+ as a result of vomiting is relatively small, considering that [K+] in gastric juice averages 15 mEq per L. Diuretic therapy for the management of accompanying hypertension and congestive heart failure is a common additional cause of potassium depletion. Within a week from the start of diuretic therapy, a mild decrease (0.3 to 0.6 mEq per L) in [K+]p occurs, and this level remains constant thereafter unless an intercurrent illness that decreases K+ intake (vomiting) or increases K+ loss (diarrhea) develops. Hypokalemia is most commonly observed with thiazides (5% of patients) than with loop diuretics (1% of patients). The decrease in [K+]p is directly proportional to the daily dosage and duration of action of the diuretic; thus, daily administration and high-dosage regimens of chlorthalidone, a long-acting thiazide, are more likely to produce severe K+ depletion and hypokalemia. The antihypertensive effect of thiazides is achieved with small dosages (6.25 to 25.0 mg daily), which have a small effect on K+ balance; consequently, high dosages of thiazides are not warranted because they will result in K+ depletion without better blood pressure control. Insulin administration in the course of treating DKA, a condition in which K+ depletion is usually present, can result in profound and symptomatic hypokalemia.
K+ Overload with Hyperkalemia. Potassium overload leading to hyperkalemia can occur because of increased K+ intake or decreased renal K+ excretion. The former occurs when the adaptive increase in renal K+ excretion is insufficient to match the larger-than-normal K+ intake. Salt substitutes are K+ salts (KCl) that mimic the taste of NaCl and their use may lead to hyperkalemia. As these products are available over the counter, patients frequently use them whether they are recommended by physicians or not. A low NaCl intake reduces the ability of the kidney to excrete K+; simultaneous ingestion of salt substitutes (K+ salts) can lead to hyperkalemia owing to the combination of increased K+ intake and reduced renal K+ excretion. In fact, a low NaCl intake is the single most commonly observed contributing factor in the development of hyperkalemia in clinical practice. Removing the salt restriction promotes increased kaliuresis, which might partially or fully correct the hyperkalemia.
Diabetes mellitus commonly damages the renal mechanisms of potassium excretion.
34 Such abnormality might result from decreased GFR, decreased tubular secretion of K
+, hypoaldosteronism or pseudohypoaldosteronism, or drugs. In the absence of generalized renal failure, a diminished renal K
+ excretion reflects either a defect in the renin-angiotensinaldosterone axis or renal resistance to aldosterone.
Renin deficiency leads to a low plasma aldosterone level that might reduce renal K+ excretion. It occurs in certain physiologic states (advanced age, expansion of ECF volume), with the use of various drugs (β-adrenergic blockers, inhibitors of prostaglandin synthesis, methyldopa), with certain toxins (lead), in some systemic diseases (diabetes mellitus), and in some renal diseases (obstructive uropathy, interstitial nephritis). A common cause of renin deficiency is the so-called type 4 renal tubular acidosis, which is characterized by impaired excretion of both K+ and H+. Perhaps its most common presentation is in elderly diabetic patients. Angiotensin-converting enzyme (ACE) inhibitors lead to hypoaldosteronism, which reduces renal K+ excretion and can increase [K+]p .
One or more of the various syndromes of diminished aldosterone activity may be observed in diabetes mellitus. A diminished aldosterone activity can occur as a result of:
1. A primary defect in the adrenal synthesis of aldosterone owing to a disease or defect in the adrenal cortex.
2. A secondary defect in the adrenal synthesis of aldosterone owing to failure in the production, release, or action of the various components of the reninangiotensin-aldosterone axis (e.g., renin deficiency).
3. End-organ resistance to aldosterone, owing to either drugs acting on the kidney (e.g., spironolactone) or renal disease.
Drug-induced mechanisms of hypoaldosteronism leading to hyperkalemia are commonly encountered in diabetic patients. Prostaglandin synthetase inhibitors, such as the nonsteroidal anti-inflammatory drugs (NSAIDs), and cyclosporin inhibit renin secretion, producing hyporeninemic hypoaldosteronism. These drugs can also cause hemodynamically induced decreases in GFR as well as direct nephrotoxicity, thereby impairing further K+ excretion. ACE inhibitors decrease plasma levels of angiotensin II resulting in decreased levels of aldosterone. Heparin acts directly on the adrenal gland, inhibiting aldosterone secretion. Increased [K+]p can occur with heparin administration in approximately 5% of hospitalized patients.
Diabetes mellitus is also associated with end-organ resistance to aldosterone, leading to hyperkalemia, a syndrome known as pseudohypoaldosteronism. This entity can develop as a result of drug administration or renal diseases. Hyperkalemia caused by spironolactone, eplerenone, triamterene, and amiloride, collectively known as K+-sparing diuretics, exemplifies drug-induced pseudohypoaldosteronism. Renal diseases that primarily damage the renal tubules (with minor decreases in GFR), collectively known as tubulointerstitial renal diseases, elicit this hyperkalemic syndrome (e.g, obstructive uropathy).
Diabetes mellitus is the single major cause of end-stage renal disease (ESRD), and therefore commonly leads to hyperkalemia because of decreased renal function (diminished GFR). In the presence of renal insufficiency, potassium balance might be maintained within normal limits until the GFR decreases to less than 25% of normal. The ability of the kidney with decreased GFR to maintain K
+ balance depends on the development of compensatory mechanisms, collectively known as K
+ adaptation, that increase the fractional K
+ excretion (FE
K) by the kidney.
34 Because [K
+]
p might remain within normal limits in patients with only 25% of overall renal function (GFR), whereas K
+ intake remains unchanged, a fourfold increase in FE
K+ must be present. As the calculated FE
K in normal individuals amounts to approximately 10%, the estimated FE
K in a patient with this degree of renal insufficiency is about 40%.
Management of Hyperkalemia. The management of K
+ retention should be initiated even in the presence of a mild degree of hyperkalemia.
34 Several measures should be undertaken at once in patients who have high-normal [K
+]
p (i.e., 5.0 mEq per L) and a disease that predisposes to hyperkalemia, such as diabetes mellitus with renal dysfunction. Severe restriction of dietary NaCl intake should be avoided because it impairs renal K
+ excretion; dietary NaCl intake should be at least 4 g per day. Restriction of dietary K
+ must be enforced. Medications that impair the renal excretion of K
+, such as K
+-sparing diuretics, should be discontinued. Metabolic acidosis, if present, should be treated with alkali therapy.
Proper management of simultaneous retention of Na+ and K+ in patients with diabetes mellitus and a salt-retaining disease (hypertension, congestive heart failure, nephrotic syndrome, renal insufficiency), who have an elevated [K+]p or even a high-normal [K+]p (i.e., 5.0 mEq per L), is effectively achieved by avoiding severe restriction of dietary NaCl intake while concomitantly administering diuretics (e.g., furosemide, thiazides). This recommendation with respect to the dietary NaCl intake should be instituted once a large ECF volume excess is no longer present. A moderate dietary NaCl intake of about 4 g per day will not result in ECF volume expansion if increased urine excretion of NaCl is achieved with the use of diuretics. This strategy secures adequate kaliuresis. Diuretics should not be administered to patients with ESRD because a meaningful kaliuretic response is not expected. A negative external K+ balance is achieved in all patients with ESRD by: (1) the utilization of cation exchange resins (such as Kayexalate, a sodium polystyrene sulfonate) that promote the excretion of K+ in the stools, and (2) dialysis (hemodialysis or peritoneal dialysis).
Hyperkalemia is the major threat to life in patients with type 4 renal tubular acidosis; therefore, the main focus of attention should be placed on correcting this electrolyte abnormality. That is the reason why dietary K+ restriction, diuretics (furosemide, thiazides), and K+-binding resins are so valuable in these patients. The intake of NaCl should be encouraged, because the availability of Na+ in the collecting tubules is a major determinant of renal K+ excretion. The administration of fludrocortisone (Florinef) in daily doses of 0.1 to 0.3 mg helps in the correction of hyperkalemia and acidosis (enhances distal acidification), yet the associated volume expansion may induce hypertension or increase its severity. Alkali therapy (1 to 2 mEq/kg/day) is useful to ensure correction of hyperkalemia and acidosis.
The following three strategies must be considered whenever severe hyperkalemia is present in any patient
34 :
1. To counterbalance the effect of hyperkalemia on the excitability of myocardial and skeletal muscle. This modality is not aimed at reducing the increased [K+]p .
2. To modify internal K+ balance, promoting the translocation of K+ from ECF to ICF. This modality will not alter the total body K+ stores.
3. To modify external K+ balance, inducing a net K+ loss from the body.
The treatment of hyperkalemia with agents that ameliorate the effects of hyperkalemia on myocardial and skeletal muscle excitability involves the administration of Ca 2+ salts (chloride or gluconate). These agents diminish tissue excitability by widening the difference between resting and threshold potentials. Calcium gluconate (20 mL of a 10% solution) can be infused intravenously over a 10-minute period. The intravenous administration of Ca 2+ salts is definitely indicated when [K+]p reaches 7.0 mEq per L or when significant electrocardiographic abnormalities (absence of P waves, prolongation of QRS complexes, etc.) are present. The effects of Ca 2+ infusion are short lasting, with peak effect noted about 5 minutes after infusion.
The most important therapeutic agent that promotes cellular K+ entry is insulin. Insulin leads to tissue uptake of K+ as well as glucose; therefore, the latter must be infused to prevent hypoglycemia in patients presenting without hyperglycemia. Considering that hyperglycemia of endogenous and exogenous origin can result in hyperkalemia (especially in diabetics), caution should be exercised as to the rate of glucose infusion. Consequently, a situation that mimics a euglycemic insulin clamp (providing enough exogenous glucose to maintain normal plasma glucose level during insulin administration) must be instituted. Additional but less important strategies that might translocate K+ from ECF to ICF are administration of ß2 agonists and infusion of sodium bicarbonate.
The first two modalities in the therapy of hyperkalemia are only temporary measures that remove the immediate threat to life resulting from hyperkalemia. Achievement of a net K+ loss is the most effective therapeutic modality to reduce and sustain a normal [K+]p in patients with severe and persistent hyperkalemia. Consequently, treatment of severe and persistent hyperkalemia must combine all three modalities.
The presence of associated clinical and laboratory abnormalities can prevent use of one or more treatment modalities of hyperkalemia. The use of K+ exchange resins by oral or rectal routes is contraindicated in patients with significant gastrointestinal symptoms. Calcium infusions are contraindicated in patients with hypercalcemia. Sodium bicarbonate infusions are contraindicated in patients with alkalemia, patients with high [HCO3˜]p , those with hypernatremia, or in patients at a significant risk of developing pulmonary edema or with significant ECF volume expansion. Severe hyperkalemia accompanied by preserved renal function usually can be corrected without dialysis. Potassium removal can be achieved in these patients by inducing an enhanced kaliuresis with the administration of fluids containing NaCl or NaHCO3 , or both, and with the use of diuretics. The majority of patients who develop severe hyperkalemia, however, have renal failure, and dialysis is the treatment of choice.


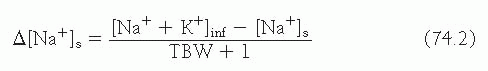

 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis



