Hereditary and familial forms of kidney cancer are encountered routinely in urologic practice. Discoveries in the genetic and molecular biology of these diseases have had a critical impact on the understanding of kidney cancer pathogenesis in nearly all subtypes of renal cortical neoplasms and their clinical features. Developing knowledge in the field has helped formulate new diagnostic and molecular therapeutic strategies for patients who have kidney cancer. This article aims to familiarize the reader with the current understanding of identified syndromes, their biology, and approaches to treatment.
Hereditary kidney cancer and familial renal cancer syndromes comprise an important group of patients in which early screening and careful follow-up of affected individuals and their relatives can positively impact disease-related morbidity and survival. Statistics on the incidence and per capita increase in cases of kidney cancer have been well documented, although not so well understood is the true proportion of patients who have familial cancers. Others have estimated that 3% to 5% of patients who have renal cancer have inherited forms of disease; however, there is evidence that a familial history of renal cancer or associated cancer syndromes may be much more common. Several factors may contribute to underreporting, including lack of available complete family history, low clinical suspicion at the time of evaluation, and the limited current understanding of genetically linked cancer syndromes.
Hereditary renal cancers (HRC) may be defined broadly as the finding of a single cancer or constellation of tumors in more than one first- or second-degree family member that may be passed on through germline mutations. Hereditary kidney cancer syndromes that have been identified with known gene mutations include von Hippel-Lindau disease (VHL), hereditary papillary renal cancer (HPRC), Birt-Hogg-Dubé syndrome (BHD), hereditary leiomyomatosis renal cell carcinoma syndrome (HLRCC), and tuberous sclerosis complex (TSC). Several other presumed heritable tumor groups have been observed but no direct mechanistic evidence has been discovered, as in patients who have familial renal oncocytoma, multilocular cystic nephroma and lymphoma. Other hereditary cancer syndromes with increased risk of associated kidney cancer also have been reported, and it is likely that more may exist.
Improved techniques of genetic research have facilitated the relatively recent discovery of the mutations associated with many HRC syndromes. Before identification of the genes responsible, heritable kidney cancer was identified based on the constellation of clinical findings used to define the phenotypic manifestations of the syndrome. These findings still provide the best initial means for evaluating individuals suspected of having hereditary kidney cancer and for screening their relatives.
Screening approaches have been described for many of the known HRC syndromes based on the study of families that have these diseases and the ages at which relevant phenotypic expression may be identified. Effective evaluation and treatment is approached best with a multidisciplinary team of physicians and nurses aware of the syndrome and it management. Often, frequent testing and imaging studies need to be coordinated between services, as do medical and surgical interventions when indicated. Combined approaches to care may help limit the risks to patients associated with the requirements of management, including radiographic exposure and surgical procedures.
Genetic testing, although available and highly reliable, must involve careful discussion of the many factors and consequences that come into play and should be performed by a trained genetic counselor. Many patients are intensely interested in pursuing genetic testing when there is a suspicion of a hereditary basis for their disease, but they may not be fully aware of the ethical and legal issues that can be raised. These issues can include the duty to inform relatives, the impact on spousal relationships within the family, and prenatal and infant testing. Many have raised concerns about discrimination in employment or insurance on the basis of a genetically inherited disease, prompting the passage of the Genetic Information Non-discrimination Act by the U.S. Congress in 2007. The expansion of the understanding of the human genome and the search for genetic causes for disease are promising but also are complex and challenging for both the individual and society.
Investigations of patients who have clinical manifestations of HRC syndromes have helped identify genes and associated germline mutations that result in cancerous and benign neoplasms in a variety of organ systems. Variable phenotypic expression within families has been recognized, although the mechanism remains unclear; it is speculated that possible environmental influences or epigenetic mechanisms are at play. Importantly, the finding of cancer genes related to the formation of seemingly unrelated tumors in separate organ systems has helped shed light on the pathogenesis of cancer in a variety of tissues and the common pathways they share.
Although familial forms of kidney cancer affect a limited group of patients, the investigation of these syndromes has had valuable impact on the management of renal cancer. For most major subtypes of renal cortical tumors there is an associated heritable form of the disease identified with a familial cohort. Screening of affected families and careful follow-up has provided a greater understanding of the natural history of these tumors, allowing insight into growth and other characteristics of the different forms of kidney cancer. Somatic mutations in these same genes have been found responsible for a large percentage of sporadic kidney cancers as well, forming the basis for an emerging understanding of the molecular pathogenesis for kidney cancer.
Hereditary kidney cancer syndromes
von Hippel-Lindau Disease
In 1895, Dr. Eugen von Hippel, an ophthalmologist, described a 23-year-old patient who suffered progressive ocular angiomas causing blindness in both eyes. Years later, at the age of 47 years, this same patient developed notable neurologic progression of his disease and died; autopsy findings were reported by Brandt. This report identified brain and ocular hemangioblastomas and pancreatic and renal cysts. Lindau, a Swedish pathologist, later linked the findings of brain, spinal, and ocular hemangioblastomas as a single disease entity that could also be associated with pancreatic and renal cysts and tumors, which he reported in 1926. The von Hippel-Lindau syndrome was named by Davison in 1936, who described the many manifestations of the disease. The syndrome includes several neoplasms, including brain and ocular hemangioblastomas, endolymphatic sac tumors of the ear, conventional clear cell renal cancer and renal cysts, pheochromocytoma, pancreatic neuroendocrine tumors and cysts, and cysts in the epididymis of the testis in men and of the broad ligament in women. In isolated cases pulmonary cysts have been seen, but as yet these cysts are not considered a part of the syndrome.
Genetics of von Hippel-Lindau disease
Genetic studies have identified the VHL gene located on the short arm of chromosome 3 (3p26-25). The VHL gene and its product (pVHL) function as a tumor suppressor gene through the regulation of hypoxia-inducible factor 1-alpha (HIF1α) and hypoxia-inducible factor 2-alpha (HIF2α). The VHL protein stabilizes HIF1α and HIF2α by forming a complex with elongin B and C in association with cullin-2. Together the pVHL/elongin BC/cullin-2 complex binds to HIF1α and HIF2α and aid in targeting the bound HIF molecule for ubiquitin-mediated proteosomal degradation. Under conditions of hypoxia, or when pVHL is dysfunctional because of mutation (pseudohypoxia), HIF is allowed to regulate several proteins involved in the pathways of angiogenesis and proliferation, including vascular endothelial growth factor, transforming growth factor alpha, and platelet-derived growth factor bets. Mutations in VHL lead to dysregulation and overaccumulation of HIF1α and HIF2α, causing activation of pathways for neovascularization and cellular proliferation in target tissues.
Inheritance of VHL is an autosomal dominant germline mutation resulting in a heterozygous, highly penetrant form of disease affecting 1 person in 36,000. The development of a second mutation causing deletion of the normal VHL allele leads to tumor formation in affected individuals. More than 300 different types of mutations have been described that involve the three exons of the VHL gene. Differences in the phenotype associated with VHL mutations have been identified and are thought to be related to the location and type of mutation. Clustering of certain tumor types from variations in phenotypic expression have been characterized broadly into four main groups ( Table 1 ). Type 1 describes VHL kindreds who do not develop pheochromocytomas. In type 2 families, pheochromocytomas are more common; this category may be subdivided further into three subtypes: 2a has low risk for renal cancer, 2b has a high risk for renal cancer, and 2c has pheochromocytoma only. It is unclear how these subtypes relate to forms of mutations in VHL and the resultant variety of seemingly dissimilar tumor types in an isolated number of organ systems.
| VHL Group | Phenotype | Genotype Mutations |
|---|---|---|
| Type 1 | Low risk of pheochromocytoma | Deletions, truncations |
| Type 2 | High risk of pheochromocytoma | Missense substitutions |
| Type 2a | Hemangiomas, low risk of renal cell carcinoma | — |
| Type 2b | Hemangiomas, high risk of renal cell carcinoma | — |
| Type 2c | Pheochromocytoma predominant | — |
Clinical features
All individuals who inherit VHL gene mutations express some form of the phenotype, although significant variation in the severity of disease can be seen even in families who share the same mutation. Earliest manifestations, particularly ocular and brain tumors, can be seen in children and infants. By age 25 years, most affected individuals have a detectable lesion. Multiorgan involvement often requires the attention and coordination of clinical care by a number of specialties.
Screening and follow-up regimens for patients who have VHL have been described. Ocular evaluation should be performed in neonates and followed annually. Neurologic, otologic, and endocrine evaluation of children should be initiated at age 8 years or earlier for any signs associated with central nervous system involvement or pheochromocytoma including tinnitus, vertigo, symptoms of loss of balance, palpitations, sweating, or frequent headaches. Abdominal imaging with ultrasound is recommended starting at age 8 years to evaluate the kidneys, adrenals, and pancreas. An MRI of the brain and spine is recommended at age 11 years. Routine CT scan imaging usually is initiated at about the age of 18 years and repeated as clinically indicated. When possible, efforts should be made to limit radiation exposure from frequent imaging studies.
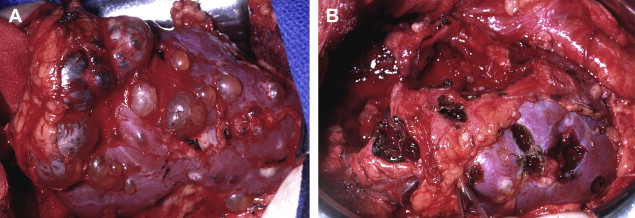
Although ocular and central nervous system hemangiomas have been the most common finding and cause of morbidity for patients who have VHL, kidney cancer has been the leading cause of death. Renal lesions, found in 40% of patients who have VHL, typically are multifocal and bilateral with cystic and partly cystic lesions detected ( Fig. 1 ). Management strategies focus on preservation of renal function and limiting intervention until solid tumors reach 3 cm in diameter on imaging studies. Nephron-sparing surgery at the time of surgical intervention involves the removal of all appreciable lesions from the kidney with priority given to lesions containing solid components, although an attempt also should be made to remove more simple-appearing cystic lesions ( Fig. 2 ). Because patients are at lifetime risk for forming tumors, repeat procedures commonly are necessary and hold a greater likelihood of complications, although reasonable functional outcomes can be obtained. Minimally invasive approaches including laparoscopic and robotic partial nephrectomy for multiple tumors and percutaneous ablation procedures have been described with favorable results at experienced centers. Long-term outcomes with regard to efficacy and renal function have not been reported.
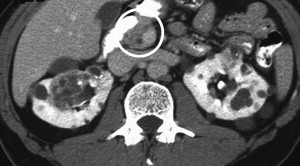
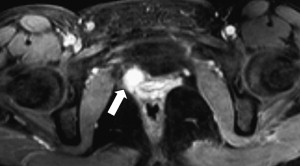
Pheochromocytomas are detected in type 2 forms of the disease and can pose a significant threat to patients if unrecognized, particularly during surgery for hemangioblastomas where there is risk of severe bleeding. Functional adrenal studies should be performed annually in all patients who have VHL, and it is recommended that meta-iodobenzylguanidine studies be performed in patients who have positive functional studies because of the possibility of extra-adrenal paragangliomas: these tumors may be multiple and reside in atypical sites including the paravesicle space ( Fig. 3 ). Surgical excision using open or laparoscopic techniques typically is performed with perioperative adrenergic blockade. Pheochromocytomas involving the adrenal gland may be resected safely using partial adrenalectomy approaches to spare a portion of the normal adrenal tissue and prevent the need for lifelong replacement steroid therapy.
Hereditary Papillary Renal Carcinoma
Genetics of hereditary papillary renal carcinoma
Type 1 HPRC has been linked to an activating mutation in the c-Met oncogene on chromosome 7 (7q31). Tumors from patients who have heritable and sporadic forms of type 1 papillary renal tumors have demonstrated this defect in c-Met, which causes aberrant activity of the intracellular tyrosine kinase domain of the membrane-bound c-Met receptor, producing the cascade of activation of the hepatocyte growth factor/c-Met pathway. Germline defects in c-Met have been identified in affected individuals; these defects may be passed on in an autosomal dominant pattern with variable penetrance, requiring trisomy of chromosome 7 with duplication of the mutated parental allele.
Clinical features
HPRC was described initially in 1994 with further features of the disease discovered through screening and clinical studies of affected kindreds. Patients inheriting the mutation show a variable degree of penetrance exhibited by the formation of renal tumors that often are multifocal and bilateral. As far as has been determined, the formation of papillary renal tumors is the only phenotype associated with the mutation. Median age of survival for affected members of these families was 52 years. Tumors often were discovered incidentally by radiographic screening.
Management of patients who have HPRC includes genetic counseling and screening within families because of the often insidious nature of these occult tumors. Preferred treatment is surgical excision with nephron-sparing approaches when possible, recognizing that these tumors often are multifocal. There are data suggesting that expectant management for HPRC tumors, allowing growth up to 3 cm in diameter before resection, does not jeopardize long-term survival or risk of metastases.
Birt-Hogg-Dubé Syndrome
Hornstein and Knickenberg were the first to describe a syndrome of perifollicular skin lesions of the face and trunk in three first-degree relatives from two generations suggesting an inherited dermatologic disorder associated with colonic polyposis. In 1977, 2 years after this earlier report, Drs. Birt, Hogg, and Dubé presented their work on dermatologic findings in 15 of 70 family members who developed skin nodules after age 25 years characterizing a genodermatosis consisting of fibrofolliculomas, trichodiscomas, and acrochordons. More recently, the cutaneous findings described by Hornstein and Knickenberg and by Birt and colleagues have been shown to reflect a spectrum of a single dermatopathology resulting from abnormal deposits of folliculin, the BHD gene product. Other associated findings now include renal tumors and pulmonary cysts or blebs that can cause spontaneous pneumothoraces. A host of other disease processes also have been encountered in patients who have BHD, although inconsistent data have failed to prove an association with the known mutation. These entities include multinodular goiter, medullary thyroid carcinoma, parotid oncocytoma, and colonic polyposis.
Genetics of Birt-Hogg-Dubé syndrome
Linkage studies have identified and localized the BHD gene to chromosome 17 (17p12q11) with inheritance demonstrating an autosomal dominant pattern. The BHD gene codes for the folliculin protein which becomes abnormally truncated and accumulates in certain tissues of patients who have BHD gene mutations. Folliculin has been proposed to take part in regulation of the mammalian target of rapamycin pathway through recently described folliculin-interacting protein and 5′-AMP–activated protein kinase. BHD normally is found in a variety of organs including brain, parotid gland, lung, pancreas, breast, prostate, kidney, and skin. Absence of BHD mRNA in renal tumors from patients who have the syndrome supports its role as a tumor suppressor gene.
Clinical features
Before the identification of the gene, diagnosis of BHD was made on clinical findings alone. The classic triad of skin fibrofolliculomas, pulmonary cysts, and renal tumors, as characterized in original screening studies of multigenerational families, defined the phenotype. Zbar and colleagues reported on 223 screened members of 33 families with the disease, identifying 98 affected individuals with clinical findings and 13 haplotype carriers. Of the 111 persons who had genetic mutations in BHD , 14% had renal tumors, compared with 2% of the nonaffected family members. Spontaneous pneumothoraces, seen predominantly in younger family members (< 40 years), were encountered in 23% of individuals who had BHD and in 1% of nonaffected family members. Only a subset of patients were evaluated with high-resolution lung CT scans, which identified lung cysts in 83% of family members who had BHD and in 10% of nonaffected relatives. Importantly, colonic polyps, which had been reported as part of the syndrome, were evaluated and found to have a statistically similar prevalence among family members with and without BHD. A more recent study in 50 new syndromic families demonstrated cutaneous manifestations in 90% of the family members who had BHD and renal tumors in 34% of affected relatives.
Renal tumors found in BHD are not as uniform as those encountered in other HRC syndromes. Tumor histologies include chromophobe tumors, oncocytoma, clear cellcarcinoma, and hybrid oncocytic tumors composed of elements of oncocytoma and chromophobe tumors. The approximate distribution of these cancers was evaluated in 130 tumors from 30 confirmed cases: 50% were hybrid oncocytoma/chromophobe tumors, 34% were chromophobe tumors, 9% were conventional clear cellcarcinoma, 5% were oncocytoma, and 2 5 were papillary tumors. Discordant histology was identified in 80% of patients, with a majority occurring as multifocal and/or bilateral tumors. Mean age at diagnosis in this cohort was 51 years.
Although clinical series suggest that most renal tumors may occur in older patients, significant tumors have been reported in younger patients. A locally invasive oncocytic tumor has been described in a 35-year-old man who developed subsequent pulmonary metastases of similar histology, and clear cell carcinoma has been resected from a patient at age 20 years. Leter and colleagues described a 39-year-old patient who had BHD and a renal tumor of mixed histology, including clear cell components, which developed distant progression resulting in death. These examples emphasize the heterogeneity of renal tumors encountered in BHD that may belie the indolent characteristics often attributed to the oncocytomas and chromophobe renal cancers associated with the disease. Untreated local tumor growth, even for more benign histologies, can result in progression to renal insufficiency and failure. For these reasons, tumors in patients who have BHD should be evaluated closely with nephron-sparing approaches initiated when possible. Intermediate data suggest that, as in patients who have VHL and HPRC, these tumors may be observed safely up to a size of 3 cm before intervention.
Hereditary Leiomyomatosis Renal Cell Carcinoma
HLRCC is a rare inherited disorder in which affected individuals are at risk of developing cutaneous and uterine leiomyomas and renal cell carcinoma. Affected HLRCC kindreds are characterized by germline mutation of the Krebs cycle enzyme, fumarate hydratase (FH). Recent studies have suggested that a poorly defined but particularly lethal form of renal cell carcinoma is associated with HLRCC. These tumors initially were described histologically as papillary neoplasms but, because of their aggressive clinical behavior, were classified separately as type 2 papillary tumors or were potentially misclassified as collecting duct tumors. With further experience and more refined characterization of the pathologic features, this nomenclature has been abandoned at experienced centers, and the term “HLRCC renal tumors” is used now.
Genetics of hereditary leiomyomatosis renal cell carcinoma
Genetic alteration in HLRCC has been mapped to a region on chromosome 1 (1q42.3-43) containing 10 exons encoding for FH. Germline mutations in the gene may be inherited in an autosomal dominant fashion. Phenotypic expression shows high penetrance for cutaneous leiomyomas and uterine fibromas, leading many early investigators to refer to the syndrome as “multiple cutaneous and uterine leiomyomatosis” or “multiple leiomyomatosis.” The occurrence of renal tumors among patients who have the syndrome has been estimated at 2% to 21%, a prevalence low enough to prompt some researchers to continue to differentiate between multiple leiomyomatosis and HLRCC although they represent the same disease. In the syndrome of HLRCC, loss of heterozygosity reveals the tumor suppressor function of the gene, demonstrating impaired enzymatic function of FH with resultant effects in Krebs cycle catabolism. Biallelic loss of FH in several models indicate that aberrant signaling may be mediated through HIF-dependent pathways, suggesting that mechanisms of pseudohypoxia may play a role in tumorigenesis in a similar fashion to VHL. Reports of clear cell histology with FH mutation in a patient who had HLRCC may support a common mechanism of tumorigenesis through HIF activation.
Clinical features
Clinical features of this disease are notable for uterine leiomyomas (fibroids) of significant severity for which, in a recently described series in the United States, as many as 89% of affected women underwent hysterectomy, 44% of them before the age of 30 years. Isolated cases of uterine leiomyosarcomas have been reported. (Ylisaukko 2006) Most patients who have the syndrome harbor some form of cutaneous leiomyoma, although these lesions may be subtle and difficult to identify. These features of cutaneous lesions and a striking familial history for highly symptomatic uterine fibroids are helpful for clinical screening of patients before considering genetic testing that may be impractical or unreliable.
Patients who have renal tumors may exhibit an aggressive, rapidly progressive disease with evidence of advanced stages of metastases despite small primary tumor size. The National Cancer Institute reported a series of 19 surgically managed patients who had HLRCC renal tumors; the median age was 39 years. Four of seven patients who had T1 tumors (≤ 7.0 cm) had regional lymph node involvement at the time of initial surgery, and 9 of 19 (49%) had evidence of metastases at the time of initial diagnosis. Based on these observations, careful evaluation and regular follow-up, including annual abdominal imaging studies, is required for patients who have HLRCC. Suspicious renal lesions may appear partly cystic and need more frequent imaging evaluation. Interval growth or change in character may require intervention. The role of nephron-sparing surgery has been questioned in this setting, emphasizing that any surgical approach must be undertaken with care. Preoperative positron emission tomography scans may prove beneficial when lymph node or nonlocalized disease is suspected.
Tuberous Sclerosis
An autosomal dominant disorder with renal manifestations, tuberous sclerosis affects as many as 1 person in 6000. TSC typically is characterized by seizures, mental retardation, and the development of hamartomas in multiple organs as a result of mutations in either the TSC1 or TSC2 gene. The gene products hamartin and tuberin together form a protein complex that inhibits activation of the downstream pathways of mammalian target of rapamycin. Aberrant function of the hamartin-tuberin complex through mutation reveals their role as tumor suppressor genes.
TSC1 has been mapped to chromosome 9 (9q34), and TSC2 is found on chromosome 16 (16p13.3). Similar to other heritable syndromes, TSC may express a spectrum of severity depending on the type and location of the mutation. Differences in the phenotype have been linked to mutations in either TSC1 or TSC2 with more severe manifestations of the syndrome, including mental retardation and renal lesions, highly associated with mutations in TSC2 . Renal manifestations seen in TSC include renal angiomyolipomas, cysts, and clear cell carcinoma. In a series of 490 patients reported by Sancak and colleagues, TSC2 mutations were significantly associated with renal cysts, angiomyolipomas, and mental retardation. Rakowski and colleagues reported on renal manifestations seen in 167 patients who had TSC and noted the presence of renal lesions in 58% of these patients. Of the patients who had renal lesions, 85% had angiomyolipomas, 45% had cysts, and 4% had clear cell carcinoma.
Related posts:
 Pathologic Features of Renal Cortical Tumors
Pathologic Features of Renal Cortical Tumors
 Epidemiology, Clinical Staging, and Presentation of Renal Cell Carcinoma
Epidemiology, Clinical Staging, and Presentation of Renal Cell Carcinoma
 Molecular Imaging of Renal Cell Carcinoma
Molecular Imaging of Renal Cell Carcinoma
 Renal Tumor Natural History: the Rationale and Role for Active Surveillance
Renal Tumor Natural History: the Rationale and Role for Active Surveillance
 Choice of Operation for Clinically Localized Renal Tumor
Choice of Operation for Clinically Localized Renal Tumor
 Lymph Node Dissection in the Management of Renal Cell Carcinoma
Lymph Node Dissection in the Management of Renal Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


