of the Na-K-ATPase pump in the skeletal and heart muscle, epithelial cells of the kidney and bladder, as well as liver and fat cells.17 This results in a series of intracellular events leading to hyperpolarization of cell membranes.17 The time course for this interaction is consistent with both an increase in enzyme activity as well as the rapid recruitment of Na-K-ATPase pumps to the cellular membrane. In contrast, chronic stimulation by insulin probably increases the total number of available pump sites. This occurs through the regulation of the Na-K-ATPase pump at the transcriptional and posttranscriptional levels by inducing the synthesis of new α and β subunits.1 McDonough and Youn,18 using a potassium clamp, have recently shown that after 10 days of potassium deprivation in rats Na-K-ATPase activity decreased by more than 50% and insulin-mediated potassium shift decreased by 94%, whereas in rats deprived of potassium for only 2 days the number of pumps did not decrease, but insulin-mediated potassium shift decreased by 80%. This would indicate that insulin resistance precedes a decrease in the number of pump expression during hypokalemia. The molecular mechanism underlying this response, however, remains poorly understood.19 Several in vitro studies, including one study in humans, have shown that insulin-driven potassium uptake by both muscle and the liver is independent of glucose uptake.15,20
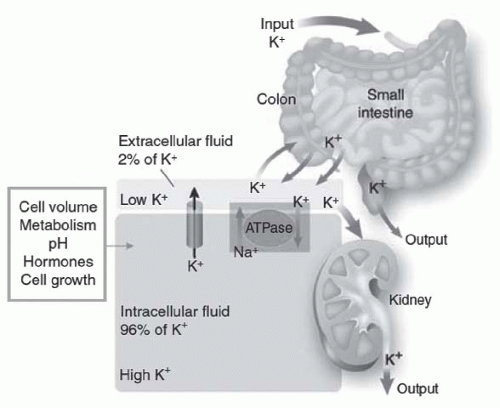 FIGURE 72.1 The distribution of potassium (K) in the body. Potassium is primarily located in cells (96%), with distribution controlled by a pump-leak mechanism involving both Na-K-ATPase and membrane potassium channels. The kidneys excrete more than 90% of the daily potassium load, and the intestines excrete the rest. (From Giebisch G, Krapf R, Wagner C. Renal and extrarenal regulation of potassium. Kidney Int. 2007;397, with permission.) |
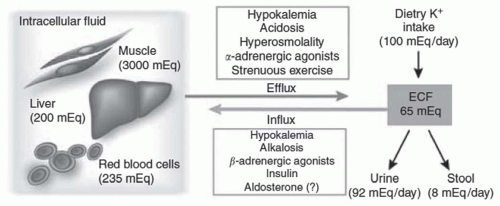 FIGURE 72.2 The distribution of potassium (K) between the intracellular and extracellular fluid compartments. Potassium distribution between the intra- and extracellular fluid is controlled by a pump-leak mechanism involving both Na-K-ATPase and membrane potassium channels. The factors noted in the figure drive potassium into or out of cells. (From Giebisch G, Krapf R, Wagner C. Renal and extrarenal regulation of potassium. Kidney Int. 2007;397, with permission.) |
with continued infusion, fell and remained lower than baseline. Other investigators have shown increased potassium tolerance in animals infused with pharmacologic doses of epinephrine22,23 despite a pancreatectomy or nephrectomy.24 Brown and coworkers25 have shown that the infusion of stress-level doses of epinephrine resulted in a decrease in serum potassium by 0.4 to 0.6 mEq per liter. Because epinephrine inhibits the renal excretion of potassium,26,27 the decline in potassium concentration is entirely accounted for by enhanced cellular potassium uptake.
for each 0.1 U change in serum pH, the serum potassium changes in the opposite direction by 0.6 mEq per liter. However, the relationship between serum potassium and serum pH is much more complex and depends on the type and severity of the acid-base disorder, the anion accompanying hydrogen, the duration of acidosis, changes in plasma bicarbonate concentration independent of changes in pH and the extent of intracellular buffering, and renal adaptation as well as hormonal changes in response to the disorder.54 In addition, in clinical settings, there are often other physiologic and pathophysiologic processes that may be present, which would affect both transcellular as well as the renal and extrarenal handling of potassium. The following generalizations should therefore be used with caution.
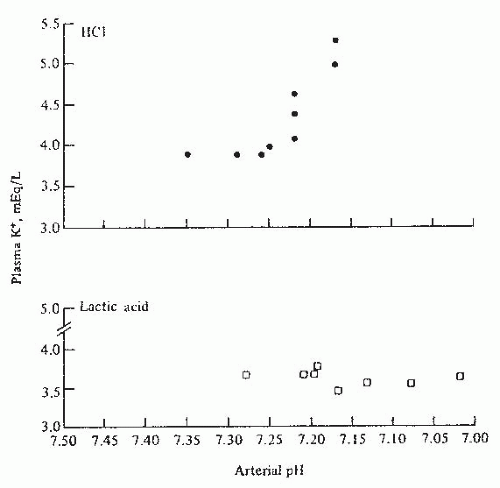 FIGURE 72.3 The effect of arterial pH on plasma potassium concentration in experimentally induced mineral acidosis (hydrochloric acid-HCl) and lactic acidosis in dogs. (From Perez GO, Oster JR, Vaamonde CA. Serum potassium concentration in acidemic states. Nephron. 1981;27:233, with permission.) |
renal function facing sudden hyperglycemia.71 These clinical observations support an independent role of sudden osmolar shifts in the regulation of serum potassium.
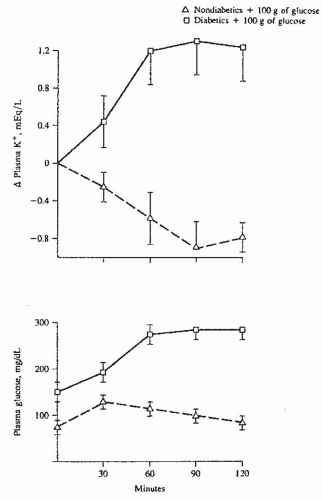 FIGURE 72.4 The effect of glucose infusion on plasma potassium and glucose concentrations in diabetics (squares) and normal subjects (triangles). The plasma potassium rises in diabetics owing to the development of hyperosmolality (hyperglycemia) but falls in normal subjects as a result of the glucose-induced release of endogenous insulin. (From Nicolis GL, Kahn T, Sanchez A, et al. Glucose-induced hyperkalemia in diabetic subjects. Arch Intern Med. 1981;141:49, with permission.) |
1.5 to 6.0 mg per deciliter) not receiving drugs that interfered with potassium homeostasis, 55% were noted to have hyperkalemia (K+ ≥ 5.0 mEq per liter).80 Treatment with drugs that interfere with potassium handling would be expected to further increase the development of hyperkalemia (see the following). Serum potassium rises with decreasing GFR; however, it often remains within normal range with GFR above 40 mL per minute.75 In this study, the rate of hyperkalemia ([K+] > 5.0) was 17% and was primarily limited to patients with CKD stage 4 and 5. However, under certain conditions, hyperkalemia may occur in patients with mild-to-moderate renal failure (Table 72.1). In a longitudinal study of patients with CKD, hyperkalemia ([K] > 5.5] was reported in only 8% of patients and, surprisingly, hypokalemia ([K] < 4.0) was more frequently seen in 15% of patients. Hypokalemia was not related to nutrition and was most likely secondary to the use of diuretics.78 This observation would indicate that electrolyte disturbances in patients with CKD are partly related to the underlying disease and partly to medications used in the management of concomitant comorbidities such as fluid overload and hypertension. However, it should be emphasized that the risk of hyperkalemia in patients with CKD, including those treated with renin-angiotensin-aldosterone system (RAAS) blockers, is relatively small.81
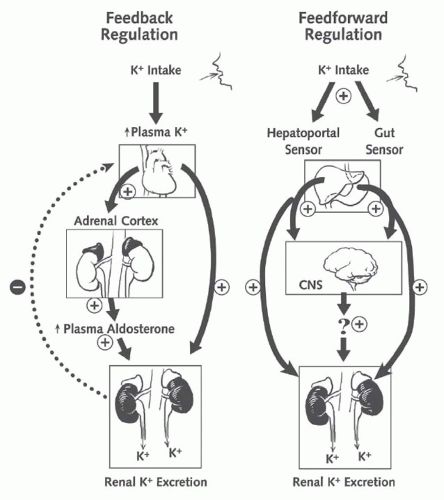 FIGURE 72.5 The integrated model of the regulation of body potassium balance: feedback and feedforward regulation. Renal potassium excretion is controlled by both feedback signals (plasma potassium concentration) and feedforward signals (liver and gut). CNS, central nervous system. (From Greenlee M, Wingo CS, McDonough AA, et al. Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med. 2009;150:619, with permission.) |
TABLE 72.1 Etiologies of Hyperkalemia in Patients with Renal Insufficiency | |||||||
|---|---|---|---|---|---|---|---|
|
liter at 60 minutes).110 The administration of glucose with potassium stimulates insulin secretion and attenuates the rise in potassium in patients on dialysis as well as normal controls.105
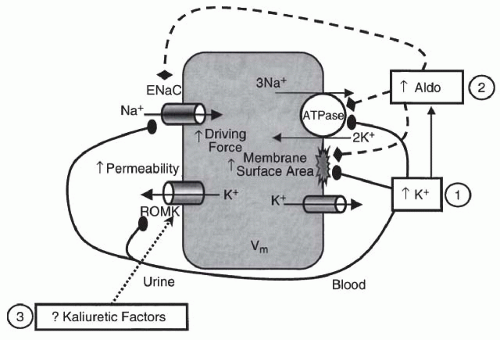 FIGURE 72.6 The major factors that regulate potassium secretion in principal cells. Sodium is reabsorbed across the luminal membrane through ENaC (epithelial sodium channels) with resultant cellular depolarization increasing the electrical driving force for potassium secretion through ROMK (potassium channels). The effects of aldosterone (Aldo) and hyperkalemia (↑K+) on potassium secretion are noted. (From Gennari FJ, Segal AS. Hyperkalemia: an adaptive response in chronic renal insufficiency. Kidney Int. 2002;62:1, with permission.) |
ammonium ion (NH4+) and bicarbonate excretion.135,136,137 Careful metabolic studies by Goodman and colleagues137 documented that patients with chronic renal failure have a daily bicarbonate deficit of approximately 13 to 19 mEq. It is notable that despite this persistent deficit, serum bicarbonate in patients with CKD after an initial drop remains stable over long periods of time.138,139 This is due chiefly to the buffering of excess hydrogen ions by bone buffers, including calcium carbonate.138
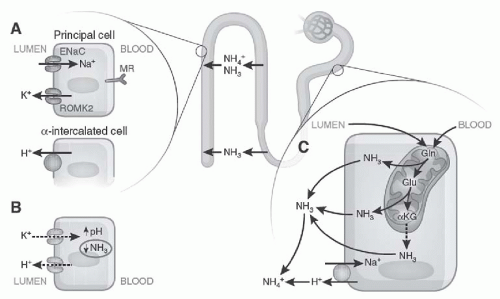 FIGURE 72.7 The factors involved in hyperkalemic acidosis. A: ENaC function at the apical surface of principal cells allows potassium secretion by ROMK (potassium channels) and the hydrogen ion by adjacent intercalated cells. B: Hyperkalemia increases intracellular pH by proton exchange, impairing the enzyme involved in ammoniagenesis. C: The process of ammoniagenesis involves deamination of glutamine, which allows ammonia to buffer the hydrogen ion in the urine. Ammonia and ammonium are reabsorbed in the medullary loop and are then excreted in the urine in the distal nephron. (From Karet FE. Mechanisms in hyperkalemic renal tubular acidosis. J Am Soc Nephrol. 2009;20:251, with permission.) |
often with HCA, our discussion is limited to the disturbances associated with renal insufficiency.
TABLE 72.2 Etiology of Chronic Hyperkalemia Due to Disturbances in Renal Potassium Excretion | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
physiologic doses of fludrocortisone. This study,155
Related posts:
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


