The electrochemical gradient for phosphorus favors movement from the intracellular to the extracellular compartment because the interior of the cell is electrically negative compared with the basolateral external surface. Therefore, the presumption has been that the exit of phosphorus across the basolateral membrane represents a mode of passive transport.321
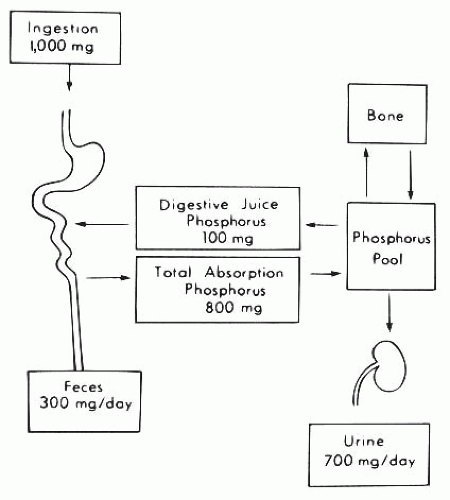 FIGURE 73.1 Summary of phosphorus metabolism in humans. Approximately 1 g of phosphorus is ingested daily, of which 300 mg is excreted in the stool and 700 mg in the urine. The gastrointestinal tract, bone, and kidney are the major organs involved in phosphorus homeostasis. |
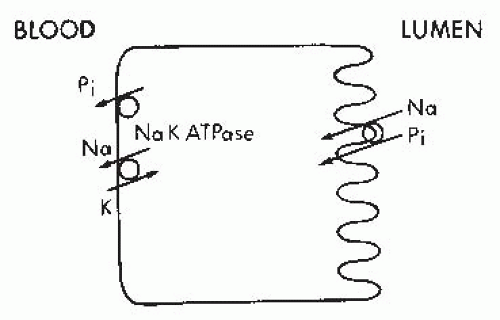 FIGURE 73.2 The apical membrane sodium-inorganic phosphate (Pi) cotransport proteins utilize the electrochemical driving force for sodium to move Pi into the cell. The electrochemical sodium gradient is maintained by active sodium extrusion across the basolateral membrane through the action of Na+-K+-ATPase. |
Nephron heterogeneity in phosphorus handling has been evaluated under a number of conditions by puncture of the papillary tip and the superficial early distal tubule, with the recorded fractional delivery representing deep and superficial nephron function, respectively. Microinjection of phosphorus tracer into thin ascending and descending limbs of loops of Henle reveals that only 80% of phosphorus was recovered in the urine, whereas 88% to 100% of phosphorus was recovered when the tracer was injected into the late superficial distal tubule. It was concluded that a significant amount of phosphorus must be reabsorbed by juxtamedullary distal tubules or by segments connecting the juxtamedullary distal tubules to the collecting ducts to account for the discrepancy between the results of superficial nephron injection and juxtamedullary nephron injections. These data support an increased reabsorptive capacity for phosphorus in deep as opposed to superficial nephrons and increased responsiveness to body Pi requirements.253,254
Drosophila septate junction protein Discs-large, and the tight junction protein ZO-1.255,288,367,596
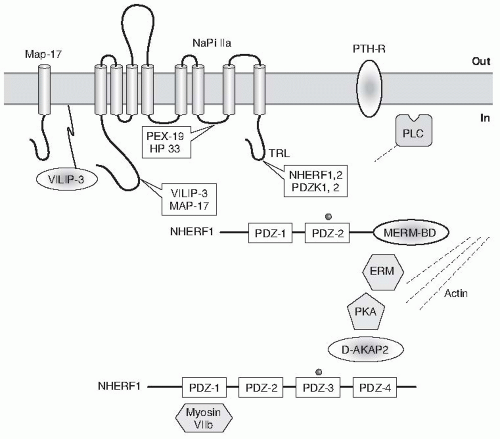 FIGURE 73.3 Interactions of NaPi-IIa with several proteins expressed in the renal proximal tubule. MAP-17 is involved in the apical location of PDZK1 (via PDZ-4) in OK cells. (From Biber J, Gisler SM, Hernando N, et al. PDZ interaction and proximal tubular phosphate reabsorption. Am J Physiol Renal Physiol. 2004;287:F871.) |
NaPi-IIa interaction with NHERF-1 was also shown in a study where targeted disruption of the mouse NHERF-1 gene was associated with decreased BBM expression and increased intracellular localization of NaPi-IIa resulting in decreased renal phosphate reabsorption and renal phosphate wasting.595 On the other hand targeted disruption of NHERF-1 did not modulate the BBM expression and localization of NHE3; however, there was impaired regulation of NHE3 in response to PKA.595
kinase A (PKA), and protein kinase C (PKC).174 The first pathway, activation of the adenylate cyclase, differs from that of PKC. Studies in OK cells show that PKA activation by PTH decreases the expression of NaPi-IIa cotransporter likely due to internalization and degradation of the transporter. Binding of PTH to its receptor leads to activation of PLC with the subsequent hydrolysis of phosphatidylinositol 4,5-bisphosphate to inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG). IP3 generation leads to the release of intracellular calcium stores. DAG activates PKC.174 In addition to its direct effect on NaPi-IIa, PTH inhibits NaPi transport indirectly by inhibiting the Na+-K+-ATPase by decreasing the favorable Na+ gradient for Pi entry into the cell.546 Recent studies indicate that the activation of PKA and PKC signaling pathways by PTH also activates mitogen activated protein (MAP) kinase (MAPK) or extracellular receptor kinase (ERK1/2) which also induces inhibition of NaPi transport.32,360
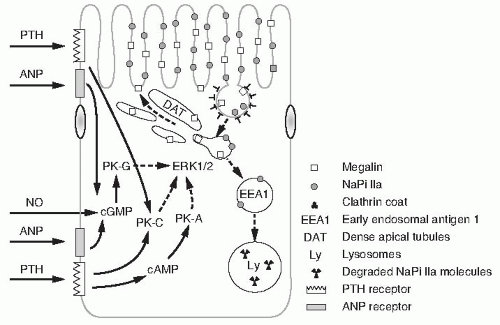 FIGURE 73.4 Mechanisms of NaPi-IIa traffic in the apical membranes of proximal tubular cells. Microvillar NaPi-IIa is retrieved in megalin containing clathrin coated vesicles. NaPi-IIa moves from the vesicles into an endosomal pool marked by EEA-1, whereas megalin is recycled through dense apical tubules back to the microvilli. The endosomal NaPi-IIa is targeted for lysosomal degradation. The process of NaPi-IIa retrieval and lysosomal degradation is stimulated by several factors (parathyroid hormone, atrial natriuretic peptide, nitric oxide) whose mechanisms of signal transduction (PK-A, PK-C, PK-G) merge in activation of ERK1/2 that modulates the process. (From Bacic D, Wagner CA, Hernando N, et al. Novel aspects in regulated expression of the renal type IIa Na/Pi-cotransporter. Kidney Int. 2004;66:S5, with permission.) |
protein levels and decreased the abundance of NaPi-IIa-specific messenger RNA (mRNA) by 31%.319 Parathyroidectomy did not affect NaPi-IIa mRNA levels. The effects of PTH were apparent within 2 hours of administration and indicate that PTH regulation of NaPi-IIa is determined by changes in the expression of NaPi-IIa protein in the renal BBMs.319
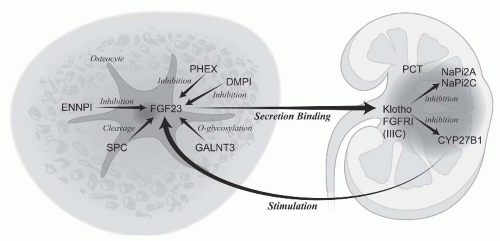 FIGURE 73.5 The skeletal-kidney endocrine axis: regulation of Pi in the exchangeable pool. FGF23 is a hormone secreted by the osteocyte to regulate proximal tubular cell Pi transport and calcitriol production. Multiple mechanisms of increased FGF23 levels cause hypophosphatemic rickets. Calcitriol stimulates FGF23 to maintain Pi levels in response to stimulation of intestinal absorption. Function of various proteins in the endocrine axis and the genetic diseases they cause are listed in Table 73.2. |
parathyroidectomized and vitamin D-depleted rats.211 A unifying interpretation of these studies was hampered by the fact that the dosages of vitamin D administered and the status of the serum calcium, phosphorus, and PTH varied considerably from study to study.
with mineralocorticoid administration, resulting in selective glucocorticoid deficiency, is associated with increased renal proximal tubular NaPi-IIa protein expression.396
significantly lower in aged rats, in spite of a significantly lower serum Pi concentration in aged rats. Thus, impaired expression of the type II renal Na-Pi cotransporter protein at the level of the apical BBM plays an important role in the age-related impairment in renal tubular reabsorption of Pi and renal tubular adaptation to a low Pi diet.635
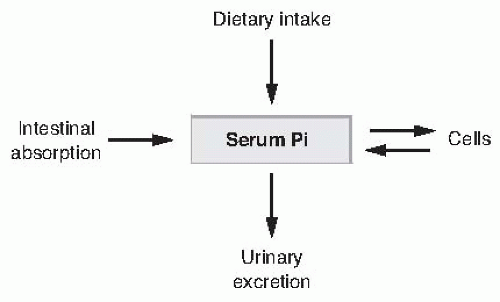 FIGURE 73.6 The major determinants of serum inorganic phosphate (Pi) concentration. |
TABLE 73.1 Causes of Hypophosphatemia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
be due to changes in the reabsorption of salt and water or to renal tubular defects specific for the reabsorption of certain solutes or phosphorus. Hypophosphatemia may also occur in the diuretic phase of acute tubular necrosis or in postobstructive diuresis, presumably due to a combination of high levels of PTH and decreased tubular reabsorption of salt and water.
TABLE 73.2 Inherited Disorders of Phosphate Homeostasis Cause Hypophosphatemic Rickets or Hyperphosphatemia and Are Components of a Bone Kidney Endocrine Axis | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
urinary losses of phosphorus that are out of proportion to the hypophosphatemia in contrast to patients with predominant phosphorus malabsorption and no secondary hyperparathyroidism in whom urinary excretion of phosphorus is low.
In vitro studies in OK cells have demonstrated that FGF23 inhibits Na/Pi cotransport activity and type IIa NaPi cotransport protein abundance by the MAPK signaling dependent pathway.725
the stimulatory heterotrimeric guanosine triphosphate binding protein, GS) in cells from affected tissues from patients with the syndrome.14,58,59,134,140,146,386,387,443,551,569,704 Kidney tissue, presumably proximal tubule, from patients has been reported to contain cells with the mutation.
the oliguric phase. During the recovery phase of acute renal failure, the combined occurrence of a profound diuresis, secondary hyperparathyroidism, and continued use of phosphate binders may lead to severe hypophosphatemia. This hypophosphatemia is usually short lived, and serum phosphorus levels return to within the normal range as the diuretic phase of acute tubular necrosis subsides.
growth. These deformities are characterized by soft loose areas in the skull known as craniotabes and costochondral swelling or bending (known as rachitic rosary). The chest usually becomes flattened, and the sternum may be pushed forward to form the so-called pigeon chest. Thoracic expansion may be greatly reduced with impairment of respiratory function. Kyphosis is a common finding. There is remarkable swelling of the joints, particularly the wrists and ankles, with characteristic anterior bowing of the legs, and fractures of the “greenstick” variety may also be seen. In adults, the symptoms are not as striking and are usually characterized by bone pain, weakness, radiolucent areas, and pseudofractures. Pseudofractures represent stretch fractures in which the normal process of healing is impaired because of a mineralization defect. Mild hypocalcemia may be present; however, hypophosphatemia is the most frequent biochemical alteration. This metabolic abnormality responds well to administration of small amounts of vitamin D.
disease seen in diabetic patients. Patients with mild degrees of hypophosphatemia are usually asymptomatic. However, if hypophosphatemia is severe—that is, if serum phosphorus levels are less than 1.5 mg per dL—a series of hematologic, neurologic, and metabolic disorders may develop. In general, the patients become anorectic and weak, and mild bone pain may be present if the hypophosphatemia persists for several months (Table 73.3).
TABLE 73.3 Clinical and Biochemical Manifestations of Marked Hypophosphatemia | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
movements in phosphate-depleted tissues is yet to be completely resolved.
When the urinary phosphorus excretion level is high, the differential diagnosis includes hyperparathyroidism, a primary renal tubular abnormality, or vitamin D-dependent or -resistant renal rickets. Measurements of serum calcium, PTH, and vitamin D and its metabolites, as well as urinary excretion of other solutes (glucose, amino acids, and bicarbonate), will usually elucidate the underlying disturbance that is responsible for the hypophosphatemia.
80% of the filtered load. This progressive phosphaturia per nephron serves to maintain the concentration of phosphorus within normal limits in plasma as renal disease progresses. The decrease in phosphate reabsorption per nephron is stimulated by increased PTH and FGF23 levels. However, when the number of nephrons is greatly diminished, if the dietary intake of phosphorus remains constant, phosphorus homeostasis can no longer be maintained and hyperphosphatemia develops. This usually occurs when the GFR falls to less than 25 mL per minute. As hyperphosphatemia develops the filtered load of phosphorus per nephron increases, phosphorus excretion rises, and phosphorus balance is reestablished but at higher concentrations of serum phosphorus, PTH, and FGF23. Hyperphosphatemia is a usual finding in patients with far-advanced renal insufficiency unless phosphorus intake in the diet has decreased through dietary manipulations or the patient is receiving phosphate binders such as calcium carbonate, sevelamer, or lanthanum carbonate that decrease the absorption of phosphate from the GI tract.568 In patients with acute kidney injury (AKI), hyperphosphatemia is a common finding.430 The degree of hyperphosphatemia in patients with acute renal failure varies considerably. It is quite marked in patients with renal insufficiency secondary to severe trauma or nontraumatic rhabdomyolysis.335 Hyperphosphatemia in CKD and AKI directly stimulates osteocyte, osteoblast, odontoblast, and vascular smooth muscle cell signaling that results in gene transcription of RUNX2 and osterix.434 In vascular smooth muscle cells of neointimal atherosclerotic plaques and cardiac valves, stimulation of RUNX2 and osterix produce matrix calcification akin to bone formation.590 In mineralizing vascular smooth muscle cells, Pi is a signal stimulating molecule, and the sodium-dependent Pi transport protein PIT1 may be the phosphorus sensing receptor.381 Thus, hyperphosphatemia is related to vascular calcification and both of these are cardiovascular risk factors in CKD.69,398 Cardiovascular mortality is extremely high in CKD and hyperphosphatemia and vascular calcification account for much of this.192,630
TABLE 73.4 Causes of Hyperphosphatemia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
paresthesias, numbness, muscle cramps, and dysphagia may be presenting symptoms.
Related posts:
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


