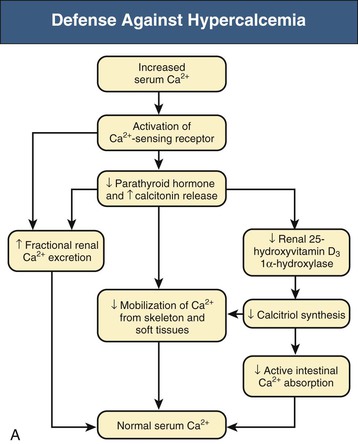
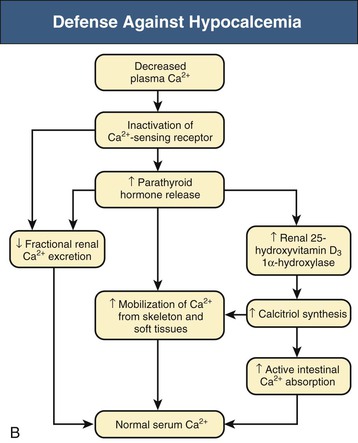
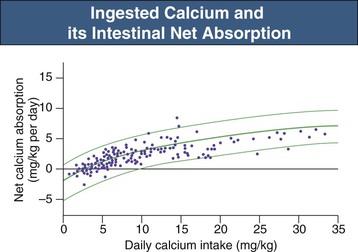
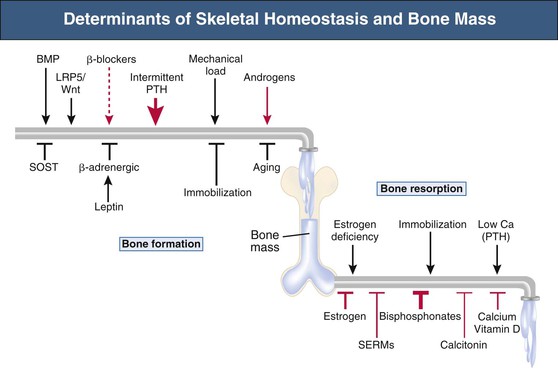
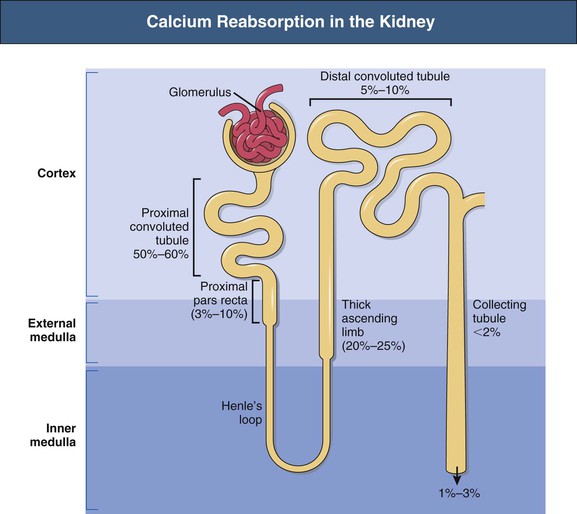
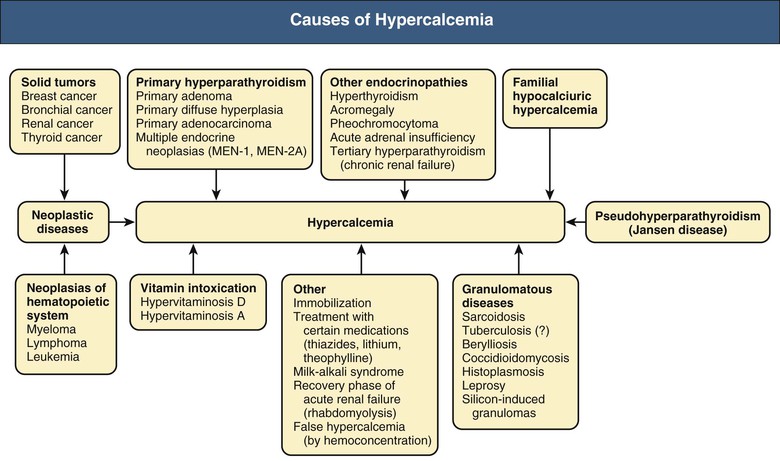
Bryan Kestenbaum, Tilman B. Drüeke Most calcium is bound and associated with bony structures (99%). The majority of free calcium, either in diffusible (ultrafilterable) nonionized form or in ionized form (Ca2+), is found in the intracellular fluid (ICF) and extracellular fluid (ECF) compartments. Similar to potassium, there is a steep concentration gradient between Ca2+ in the ICF versus ECF milieu (Fig. 10-1). The serum concentration of Ca2+ is tightly regulated within a narrow range by the actions of parathyroid hormone (PTH, parathormone) and calcitriol (1,25-dihydroxycholecalciferol). The physiologic role of other calcium regulatory hormones, such as calcitonin, estrogens, and prolactin, is less clear. Figure 10-2 demonstrates the physiologic defense mechanisms used to counter changes in serum Ca2+ levels.1 Serum Ca2+ levels are also influenced by acid-base status, with alkalosis causing a decrease in Ca2+ and acidosis an increase in Ca2+. Long-term maintenance of calcium homeostasis depends on (1) the adaptation of intestinal Ca2+ absorption to the needs of the organism, (2) the balance between bone accretion and resorption, and (3) urinary excretion of calcium (Fig. 10-3). Gastrointestinal (GI) calcium absorption is a selective process; only about 25% of total dietary calcium is absorbed. Ca2+ transport across the intestinal wall occurs in two directions: absorption and secretion. Absorption can be subdivided into transcellular and paracellular flow (Fig. 10-4). Transcellular calcium flux takes place through the transient receptor potential TRPV6 calcium channel.2 Calcitriol is calcium’s most important hormonal regulatory factor. After binding to and activating the vitamin D receptor (VDR), calcitriol increases active transport by inducing the expression of TRPV6, calbindin-D9k, and Ca2+-ATPase (PMCA1b).3 Other hormones, including estrogens, prolactin, growth hormone, and PTH, also stimulate Ca2+ absorption, either directly or indirectly. The amount of dietary calcium intake also regulates the proportion of calcium absorbed through the GI tract (Fig. 10-5).4 Cutaneous synthesis on exposure to ultraviolet (UV) light converts 7-dehydrocholesterol into vitamin D substrate (cholecalciferol). Vitamin D substrate is also obtained through the diet and supplementation. Cholecalciferol (and ergocalciferol) have minimal inherent biologic activity and require two hydroxylation steps for full hormonal activity. 25-Hydroxylation occurs in the liver, is thought to be non–rate limiting, and is widely accepted as a summary measure of vitamin D stores. Further hydroxylation to 1,25-dihydroxyvitamin D (calcitriol) occurs predominantly in the kidney; however, other tissues can also perform this hydroxylation step, in particular macrophages and parathyroid tissue. Increased calcium absorption is required in puberty, pregnancy, and lactation. In each of these states, calcitriol synthesis is increased to enhance GI calcium absorption. Intestinal Ca2+ absorption is also increased in states of vitamin D excess and acromegaly. Rarely, ingestion of calcium and alkali in large quantities can overwhelm GI checks on calcium absorption, resulting in hypercalcemia (milk-alkali syndrome). However, innate limitations on GI calcium absorption prevent this condition from occurring in most individuals. A decrease in intestinal Ca2+ transport occurs in advanced age, chronic kidney disease (CKD), gastrectomy, intestinal malabsorption syndromes, diabetes mellitus, corticosteroid treatment, and estrogen deficiency and with dietary factors such as high vegetable fiber and fat content, low Ca2+–phosphate ratio in food, and fructose ingestion. The decrease in Ca2+ absorption in older adults probably results from multiple factors in addition to lower serum calcitriol and intestinal VDR levels.5 The net balance between Ca2+ entry and exit fluxes is positive during skeletal growth in children, zero in young adults, and negative in the elderly. Exchangeable skeletal Ca2+ contributes to maintaining extracellular Ca2+ homeostasis. Several growth factors, hormones, and genetic factors participate in the differentiation from the mesenchymal precursor cell to the osteoblast and the maturation of the osteoclast from its granulocyte-macrophage precursor cell (Fig. 10-6). The regulation of bone formation and resorption involves many hormones, growth factors, and mechanical factors6,7 (Fig. 10-7). The kidneys play a major role in the minute-by-minute regulation of calcium, and the intestine and skeleton ensure homeostasis in the midterm and long term. To perform its task, the kidney uses a complex system of filtration and reabsorption (Fig. 10-8).8 Moreover, the kidney controls conversion of substrate 25-hydroxyvitamin D to calcitriol, which then regulates GI calcium absorption through the TRPV6 channel. Renal calcitriol production is stimulated by PTH and inhibited by hyperphosphatemia and fibroblast growth factor 23 (FGF-23).The adjustment of blood Ca2+ is mainly achieved by modulation of tubular Ca2+ reabsorption in response to the body’s needs, perfectly compensating minor increases or decreases in the filtered load of calcium at the glomerular level, which is normally about 220 mmol (8800 mg) in 24 hours (see Fig. 10-3). In the proximal tubule, Ca2+ reabsorption follows the convective flow of salt and water, whereas the transport mechanisms are more complex in the distal segments. States of excess volume delivery to the kidney, such as a high-sodium diet, diminish the concentration gradient between proximal tubule and peritubular capillary, reducing calcium absorption and increasing calcium in the urine. This mechanism likely plays a role in the pathogenesis of calcium-based kidney stones. On the other hand, volume depletion increases salt, water, and (by convection) calcium reabsorption in the proximal tubule, exacerbating states of hypercalcemia. For this reason, intravascular volume repletion is an essential component of the treatment for hypercalcemia. In the thick ascending limb of Henle loop (TAL), Ca2+ transport is linked with activity of the Na+-K+-2Cl− transporter. Stimulation of the Ca2+-sensing receptor (CaRG) on the basolateral membrane decreases activity of rectifying outer medulla potassium (ROMK) channels located on apical cell surfaces, impeding activity of the Na+-K+-2Cl− cotransporter. The result is dissipation of the electrochemical gradient and decreased calcium reabsorption through claudin-16 in the tight junction. In contrast, understimulation of CaRG because of lower serum calcium levels enhances ROMK activity, resulting in greater calcium reabsorption. In the distal tubule, active Ca2+ transport occurs via the transcellular route through the calcium transient receptor potential channel V5 (TRPV5) located in the apical membrane and coupled with a specific basolateral calcium-ATPase (PMCa1b) and a Na+-Ca2+ exchanger (NCX1). Both PTH and calcitriol regulate distal tubular Ca2+ transport. The expression and role of CaRG in tubular segments other than TAL has recently been questioned.9 Numerous factors control the glomerular filtration and tubular Ca2+ reabsorption.2,10,11 Elevated renal blood flow and glomerular filtration pressure (during ECF volume expansion) lead to an increase in filtered load, as do changes in the ultrafiltration coefficient Kf and an increase in glomerular surface. True hypercalcemia also increases ultrafilterable calcium; whereas true hypocalcemia decreases it. PTH decreases glomerular Kf and thus reduces the ultrafiltered calcium load; PTH increases Ca2+ reabsorption in the distal nephron. However, PTH and PTH-related peptide (PTHrp) also induce hypercalcemia, and, because of the increase in serum calcium, the excretion of filtered calcium is elevated overall. Both extracellular Ca2+ and intracellular Ca2+ reduce tubular calcium reabsorption by activating CaRG, and the effect of extracellular Ca2+ is enhanced by the calcimimetics (discussed later). Respiratory acidosis leads to hypercalciuria through an increase in plasma Ca2+, whereas metabolic acidosis leads to hypercalciuria through Ca2+ release from bone and an inhibitory effect on tubular Ca2+ reabsorption. Conversely, alkali ingestion reduces renal excretion of calcium. The enhancing effect of phosphate depletion on urinary calcium elimination can partly occur through changes in PTH and calcitriol secretion. Dietary factors modify urinary excretion of calcium mostly by their effects on intestinal Ca2+ absorption. Several classes of diuretics act directly on the tubules. Loop diuretics and mannitol favor hypercalciuria, with a major impact on TAL, whereas the thiazide diuretics and amiloride induce hypocalciuria. Increased total plasma calcium concentration (plasma [Ca2+]) can result from an increase in plasma proteins (false hypercalcemia) or from an increase in plasma ionized Ca2+ (true hypercalcemia). Only increased Ca2+ leads to clinically relevant hypercalcemia. When only the value for the total plasma [Ca2+] is available rather than the free level ions, as generally occurs in clinical practice, plasma [Ca2+] can be estimated by taking into account plasma albumin; an increase in albumin of 1.0 g/dl reflects a concomitant increase of 0.20 to 0.25 mmol/l (0.8 to 1.0 mg/dl) plasma calcium. However, simple correction of total calcium for serum albumin may not be valid in patients with CKD. A study of 691 individuals with CKD stages 3 to 5 showed that the albumin-corrected total serum [Ca2+] correlated poorly with the ionized [Ca2+].11a Moreover, the two most common assays used to measure serum albumin yield discordant results in uremic patients, the bromcresol purple method providing lower albumin values than the bromcresol green method. The Ca2+-sensing receptor has been identified in numerous tissues, and its function and role in various disease states are well defined.12 Mutations of the gene for CaRG result in various clinical syndromes characterized by hypercalcemia or hypocalcemia (see later discussion). Other Ca2+ receptors subsequently cloned include GPRC6A, which is expressed in osteoblasts and is clearly distinct from CaRG.13 Although its functional properties have been characterized, however, the role of GPRC6A in regulation of osteoblast function and in human disease is still unknown. True hypercalcemia results from an increase in intestinal Ca2+ absorption, a stimulation of bone resorption, or a decrease in urinary Ca2+ excretion. Enhanced bone resorption is the predominant mechanism in most cases of hypercalcemia (Fig. 10-9). The main cause of hypercalcemia is excessive bone resorption induced by neoplastic processes, usually solid tumors. Tumors of the breast, lung, and kidney are the most common, followed by hematopoietic neoplasias, particularly myeloma. Most hypercalcemic tumors act on the skeleton either by direct invasion (metastases) or by producing factors that stimulate osteoclastic activity, including most frequently PTHrp, as well as other factors activating osteoclasts, transforming growth factors (TGFs), prostaglandin E (PGE), and rarely calcitriol and tumor necrosis factor α (TNF-α), and even more rarely PTH, as produced by parathyroid cancer. Only 8 of the 13 first amino acids of PTHrp are identical with those of the N-terminal fragment of PTH, but the effects of both hormones on target cells are mostly the same. In addition to their common receptor, the PTH/PTHrp receptor, at least one other receptor exists, the PTH2 receptor, which recognizes only PTH, with similar or identical signal transduction systems. In pathologic conditions, most of the PTHrp in the body is synthesized by solid tumors. PTHrp stimulates osteoclastic activity and thus liberates excess quantities of calcium from the skeleton. Osteoclast-activating factors secreted by myeloma plasmocytes and the lymphoblasts of malignant lymphomas include interleukins (IL-1α, IL-1β, IL-6) and TNF-α, which stimulate osteoclast activity. Other osteoclast-activating factors include PGE1 and PGE2, which can be secreted in large amounts by some tumors, especially renal masses. Some lymphoid tumors synthesize excess quantities of calcitriol. This capacity has been described in Hodgkin disease, T cell lymphoma, and leiomyoblastoma. The second most common cause of hypercalcemia is primary hyperparathyroidism. Early diagnosis is achieved through the widespread use of routine plasma calcium determination. In more than 80% of patients, the disease is caused by adenoma of a single parathyroid gland; 10% to 15% have diffuse hyperplasia of all glands and less than 5%, a parathyroid cancer. Primary hyperparathyroidism can be inherited either as diffuse hyperplasia of the parathyroid glands alone or as a component in multiple glandular hereditary endocrine disorders. Patients with multiple endocrine neoplasia type 1 (MEN-1) have various combinations of parathyroid, anterior pituitary, enteropancreatic, and other endocrine tumors, resulting in hypersecretion of prolactin and gastrin in addition to parathormone. MEN-1 is caused by inactivating germ-line mutations of a tumor suppressor gene (MEN-1) that is inherited as an autosomal dominant trait. In MEN-2A, the thyroid medulla and the adrenal medulla are involved with the parathyroid, resulting in hypersecretion of calcitonin and catecholamines. MEN-2A is caused by activating mutations of the RET proto-oncogene. It is also inherited as an autosomal dominant trait. Not all patients with mildly elevated plasma PTH levels develop hypercalcemia. The development of the latter may depend on a concomitant elevation of plasma calcitriol. Jansen disease is a rare hereditary form of short-limbed dwarfism characterized by severe hypercalcemia, hypophosphatemia, and metaphyseal chondrodysplasia. It is the result of activating mutations of the gene coding for the PTH/PTHrp receptor, a particular form of pseudohyperparathyroidism. Familial hypocalciuric hypercalcemia (FHH) is a rare hereditary disease caused by inactivating mutations in the gene for CaRG with autosomal dominant transmission.14 The key clinical finding in FHH is an inappropriately low urinary calcium excretion in the setting of high-normal serum calcium. Other features include hypophosphatemia, hyperchloremia, and hypermagnesemia. Plasma PTH concentration is normal or moderately elevated, and the fractional excretion of calcium is lower than that observed in hyperparathyroidism. The fractional excretion of calcium is best assessed by calculating the calcium-to-creatinine clearance ratio from a 24-hour urine collection, as follows: In FHH, the urine calcium–creatinine clearance ratio (calculated using milligrams) is usually less than 0.01. The urine Ca–creatinine concentration ratio is most often less than 0.07 mg/mg (0.2 mmol/mmol). In patients with FHH, hypercalcemia never leads to severe clinical signs, except in the neonatal period, when malignant hypercalcemia can be observed in the patient with severe hyperparathyroidism. Other endocrine disorders associated with moderate hypercalcemia include hyperthyroidism, acromegaly, and pheochromocytoma. In addition, acute adrenal insufficiency should also be considered in the differential diagnosis, although in these patients, hypercalcemia is usually false and results from hemoconcentration. Hypercalcemia can also occur in severe forms of the secondary hyperparathyroidism of CKD, which have also been called “tertiary hyperparathyroidism.” However, this is relatively uncommon to date because low circulating calcitriol concentrations in CKD patients limit GI calcium absorption, and overfunctioning parathyroid glands can be controlled more easily. Several other disorders sometimes induce hypercalcemia. Among the granulomatoses, sarcoidosis results in increased plasma Ca2+, particularly in patients exposed to sunlight. The cause is uncontrolled production of calcitriol by macrophages, a result of 1α-hydroxylase in the macrophages within the granulomas. Tuberculosis, leprosy, berylliosis, and many other granulomatous diseases are occasionally (but rarely compared with sarcoidosis) the origin of hypercalcemia, probably through the same mechanism. Hypercalcemia may also result from prolonged bed rest, especially in patients with preexisting high bone turnover rates, such as children, adolescents, and patients with Paget disease. Recovery from acute renal failure secondary to rhabdomyolysis-induced renal failure has been associated with hypercalcemia in 25% of cases and likely results from mobilization of soft tissue calcium deposits and through increases in PTH and calcitriol. Other causes include intoxication by vitamin D or its derivatives, vitamin A overload, and thiazide diuretics. Large doses of calcium (5 to 10 g/day), especially when ingested with alkali (antacids), can also lead to hypercalcemia and nephrocalcinosis (milk-alkali syndrome). The severity of clinical symptoms and signs caused by hypercalcemia depends on not only the degree but also the velocity of its development. Severe hypercalcemia can be accompanied by few manifestations in some patients because of its slow, progressive development, whereas much less severe hypercalcemia can lead to major disorders if it develops rapidly. In general, the first symptoms are increasing fatigue, muscle weakness, inability to concentrate, nervousness, increased sleepiness, and depression. Subsequently, GI signs may occur, such as constipation, nausea and vomiting, and rarely peptic ulcer disease or pancreatitis. Renal-related signs include polyuria (secondary to nephrogenic diabetes insipidus), urinary tract stones and their complications, and occasionally tubulointerstitial disease with medullary and to a lesser extent cortical deposition of calcium (nephrocalcinosis). Neuropsychiatric manifestations include headache, loss of memory, somnolence, stupor, and rarely coma. Ocular symptoms include conjunctivitis from crystal deposition and rarely band keratopathy. Osteoarticular pain in primary hyperparathyroidism has become rare in Western countries because of earlier diagnosis of hypercalcemia. High blood pressure can be induced by hypercalcemia, but it is more frequently a chance association. Soft tissue calcifications can occur with longstanding hypercalcemia. The electrocardiogram (ECG) may show shortening of the QT interval and coving of the ST wave. Hypercalcemia may also increase cardiac contractility and can amplify digitalis toxicity. When the history and clinical examination are not helpful, primary hyperparathyroidism should be investigated first. Although this is only the second most frequent cause, its laboratory diagnosis is at present easier than that of tumoral involvement. The initial step should be to measure plasma PTH and total or ionized [Ca2+], as available. When the PTH value is high-normal or high in the presence of high-normal or high [Ca2+], the diagnosis of primary hyperparathyroidism is likely (although FHH is not excluded). Other helpful laboratory tests may include phosphate, creatinine, total alkaline phosphatases, and urinary calcium and creatinine. Note that prolonged hypercalcemia is often associated with (reversible) increased serum creatinine. Cervical ultrasound and sestamibi isotope scanning may be performed to locate a parathyroid adenoma, although some surgeons still consider these examinations unnecessary before an initial neck exploration. However, imaging is indispensable in patients with recurrent hyperparathyroidism and having unilateral neck surgery under local anesthesia. If the plasma PTH level is low-normal or low, the possibility of a neoplastic disorder should be seriously considered. A low serum anion gap may be caused by multiple myeloma, because occasionally the monoclonal IgG is positively charged. In addition to the usual examinations, such as serum protein electrophoresis, measurement of the plasma PTHrp level can now be done in specialized laboratories. Exogenous vitamin D overload is associated with increased serum 25-hydroxyvitamin D levels, and granulomatous diseases such as sarcoidosis are associated with elevated calcitriol levels and with increased serum angiotensin-converting enzyme (ACE) activity. Treatment of the patient with hypercalcemia is aimed at the underlying cause. However, severe and symptomatic hypercalcemia requires rapid correction, whatever the cause. Initially, the patient must be rapidly rehydrated with isotonic saline to correct the often marked volume depletion, to reduce proximal tubule calcium reabsorption and enhance calcium excretion. Only when euvolemia is established should loop diuretics be used (e.g., IV furosemide, 100 to 200 mg every other hour) to facilitate urinary excretion of calcium; however, intravenous saline should be continued to prevent hypovolemia. Oral intake and IV administration of fluids and electrolytes should be carefully monitored, and urinary and gastric excretions measured if excessive, especially potassium, magnesium, and phosphate. Acid-basic balance should also be carefully monitored. Severe cardiac failure and CKD are contraindications to massive ECF volume expansion along with diuretics. Bisphosphonates are the treatment of first choice, especially in patients with hypercalcemia associated with cancer.15 These agents inhibit bone resorption as well as calcitriol synthesis. Bisphosphonates can be administered orally (PO) in less severe disease or intravenously (IV) in severe hypercalcemia. Common bisphosphonates include clodronate (1600 to 3200 mg/day PO), pamidronate (15 to 90 mg IV over 1 to 3 days, once a month), and alendronate (10 mg/day PO). IV doses should be infused in 500 ml of isotonic saline or dextrose over at least 2 hours and up to 24 hours. Although package warnings state that bisphosphonates should be used with caution in patients with CKD, virtually no safety data support such warnings. Bisphosphonates have been safely used in CKD patients for the correction of hypercalcemia. A reasonable strategy is first to attempt correcting acute kidney injury before administering a bisphosphonate, and to avoid repetitive dosing; a single 60-mg dose of pamidronate can maintain normal [Ca2+] for weeks. Calcitonin acts within hours, especially after IV administration. Human, porcine, or salmon calcitonin can be given. However, calcitonin often has no effect, or only a short-term effect, because of the rapid development of tachyphylaxis. Mithramycin is a cytostatic drug with remarkable power to inhibit bone resorption. Administration of a single IV dose is generally followed by a rapid decline in plasma calcium within a few hours, and this effect lasts several days. However, mithramycin is reserved for malignant hypercalcemia, and its cytotoxic effect and side effects (thrombocytopenia, liver function abnormalities) preclude prolonged administration. The maximal daily dose of mithramycin is 25 µg/kg.
Disorders of Calcium, Phosphate, and Magnesium Metabolism
Calcium Homeostasis and Disorders of Calcium Metabolism
Distribution of Calcium in the Organism
Intestinal, Skeletal, and Renal Handling of Calcium
Hypercalcemia
Causes of Hypercalcemia
Malignant Neoplasias
Primary Hyperparathyroidism
Jansen Disease
Familial Hypocalciuric Hypercalcemia
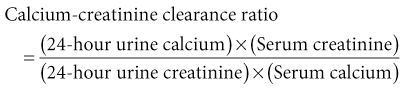
Other Endocrine Causes
Other Causes
Clinical Manifestations
Diagnosis
Treatment
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Disorders of Calcium, Phosphate, and Magnesium Metabolism
Chapter 10

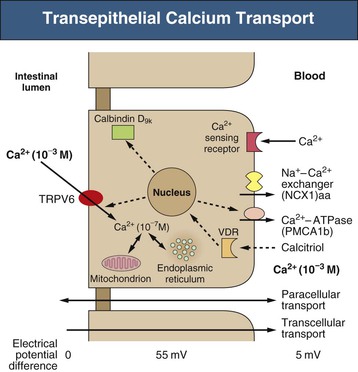
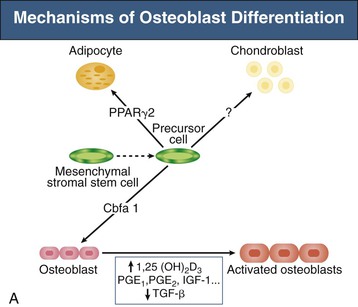
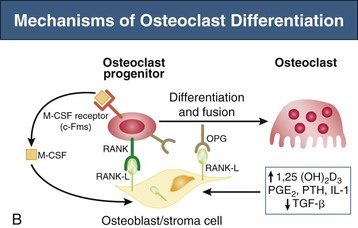
Figure 10-7 Determinants of skeletal homeostasis and bone mass. Physiologic (black) and pharmacologic (red) stimulators and inhibitors of bone formation and resorption are listed with the relative impact (represented by thickness of arrows). BMP, Bone morphogenetic protein; LRP5, low-density lipoprotein receptor–related protein 5; PTH, parathyroid protein; SERMs, selective estrogen receptor modulators; SOST, sclerostin. (From reference 6.)






