Eric Judd, Paul W. Sanders, Anupam Agarwal
Diagnosis and Clinical Evaluation of Acute Kidney Injury
The term acute renal failure (ARF) describes the clinical syndrome in which an abrupt (hours to days) decrease in renal function leads to the accumulation of nitrogenous waste products and, commonly, a reduction in urine output. Acute kidney injury (AKI) is the new consensus term for ARF.1–4 This change in terminology served to standardize a definition for the syndrome as well as incorporate new knowledge that small rises in serum creatinine (0.3 mg/dl) are associated with increased mortality and morbidity.5
The Acute Dialysis Quality Initiative first defined AKI with the RIFLE criteria (risk, injury, failure, loss, end stage) in 2004 (Table 71-1).2 The Acute Kidney Injury Network (AKIN) later supported the RIFLE criteria with minor modifications (see Table 71-1).3,4 Both definitions have been validated in large patient cohorts, and the Kidney Disease: Improving Global Outcomes (KDIGO) group merged the two definitions in a recent guideline update (Table 71-2). AKI is now defined as an increase in serum creatinine of 0.3 mg/dl or more within 48 hours of observation or 1.5 times baseline or greater, which is known or presumed to have occurred within 7 days, or a reduction in urine volume below 0.5 ml/kg/h for 6 hours.1
Table 71-1
AKIN and RIFLE classifications of acute kidney injury.
For conversion of creatinine expressed in SI units to mg/dl, divide by 88.4. For AKIN, the increase in creatinine must occur in less than 48 hours. For RIFLE, AKI should be both abrupt (within 1 to 7 days) and sustained (more than 24 hours). ESRD, End-stage renal disease; GFR, glomerular filtration rate; RIFLE, risk, injury, failure, loss, end stage; RRT, renal replacement therapy.
| Acute Kidney Injury Network (AKIN) and RIFLE Classifications of Acute Kidney Injury | |||
| AKIN Staging | Urine Output (Common to Both) | RIFLE | |
| Serum Creatinine | Class | Serum Creatinine or GFR | |
| Stage 1: Increase of ≥0.3 mg/dl (≥26.5 µmol/l) or increase to ≥150% to 200% (1.5-fold to twofold) from baseline | <0.5 ml/kg/h for >6 h | Risk | Increase in serum creatinine ×1.5 or GFR decrease >25% |
| Stage 2: Increased to >200% to 300% (more than twofold to threefold) from baseline | <0.5 ml/kg/h for >12 h | Injury | Serum creatinine ×2 or GFR decreased >50% |
| Stage 3: Increased to >300% (more than threefold) from baseline, or ≥4.0 mg/dl (≥354 µmol/l) with an acute increase of at least 0.5 mg/dl (44 µmol/l) or on RRT | <0.3 ml/kg/h for 24 h or anuria for 12 h | Failure | Serum creatinine ×3, or serum creatinine > 4 mg/dl (>354 µmol/l) with an acute rise >0.5 mg/dl (>44 µmol/l) or GFR decreased >75% |
| Loss | Persistent acute renal failure = complete loss of kidney function for longer than 4 weeks | ||
| End-stage kidney disease | ESRD > 3 months | ||
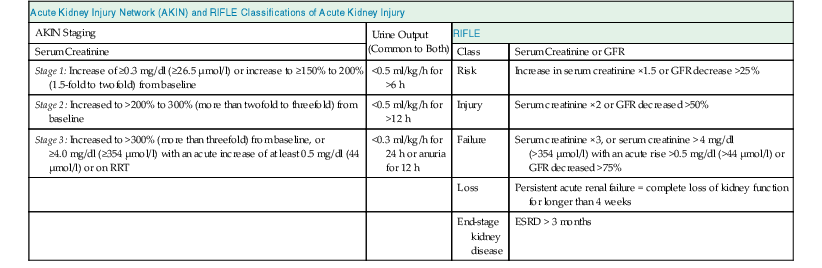
(Modified from reference 1.)
Table 71-2
KDIGO composite staging of AKI.
AKI, Acute kidney injury; eGFR, estimated glomerular filtration rate.
| Kidney Disease: Improving Global Outcomes (KDIGO) Composite Staging of AKI | ||
| Stage | Serum Creatinine | Urine Output |
| 1 | 1.5-1.9× baseline OR ≥0.3 mg/dl (≥26 µmol/l) increase | <0.5 ml/kg/h for 6-12 h |
| 2 | 2.0-2.9× baseline | <0.5 ml/kg/h for ≥12 h |
| 3 | 3.0× baseline OR Increase in serum creatinine to ≥4.0 mg/dl (≥352 µmol/l) OR Initiation of renal replacement therapy OR, in patients younger than 18 years, decrease in eGFR to <35 ml/min/1.73 m2 | <0.3 ml/kg/h for ≥24 h OR Anuria for ≥12 h |
(From reference 1.)
The incidence of AKI varies depending on the population studied and the definition used in the analysis. In a community-based cohort in northern California, from 1996 to 2003, the annual incidence of AKI in which dialysis was not required was 3841 per million population per year when AKI was defined as an increase in serum creatinine of 0.5 mg/dl or more from a baseline of below 2.0 mg/dL, or an increase of 1.0 mg/dl or more from a baseline of 2.0 to 5.0 mg/dl.6 In the United States, AKI is present in 1.9% of hospital inpatients and is especially common in critically ill patients, in whom the prevalence is higher than 60% during intensive care unit (ICU) stays.7,8 AKI severity increases in the ICU, with 5% to 6% of patients requiring renal replacement therapy, and in the United States the incidence of AKI patients who require dialysis is growing at a rate of 10% annually.9,10
The development of AKI has significant short- and long-term consequences. Despite major advances in dialysis and intensive care, the mortality rates for patients with AKI in the ICU remain high, 37% to 60%.9,11 In a large population of almost 20,000 hospitalized adults, AKI severity was directly associated with increased in-hospital mortality, lengthened hospital stay, and higher overall cost. The associations were present for changes in serum creatinine as low as 0.3 mg/dl.5 Patients with AKI who survive hospitalization also have an increase in long-term mortality, with an adjusted mortality risk of 1.4, which is amplified with increasing AKI stage.12 Furthermore, AKI survivors are at increased risk for developing comorbidities including chronic kidney disease (CKD).13
Diagnosis and Clinical Evaluation of Acute Kidney Injury
Early Detection of Acute Kidney Injury
With the knowledge that patient outcomes worsen with increasing severity of AKI beginning with small declines in renal function, research efforts have focused on early detection of AKI. Increases in serum creatinine levels currently define AKI, yet when used as a marker of renal function, serum creatinine concentrations have multiple limitations. In addition to a steady-state balance of creatinine production and excretion being required for appropriate estimation of glomerular filtration rate (GFR), serum creatinine concentrations may not rise for subtle declines in GFR and are slow to rise for rapid falls in GFR. Furthermore, the generation of creatinine from muscle is reduced in sepsis-induced AKI, and serum creatinine concentrations may not increase proportionally to GFR decline.14 A window of time exists in which kidney injury goes undetected until serum creatinine concentrations rise (8 to 48 hours) (Fig. 71-1).15 Novel serum and urine biomarkers are undergoing investigation as potential early indicators of AKI. These biomarkers, which include kidney injury molecule 1 (KIM-1), neutrophil gelatinase–associated lipocalin (NGAL), cystatin C, interleukin (IL)–18, and others, not only offer the potential for early detection of AKI but also may allow for improved prognostication and insight into the specific cause of AKI.
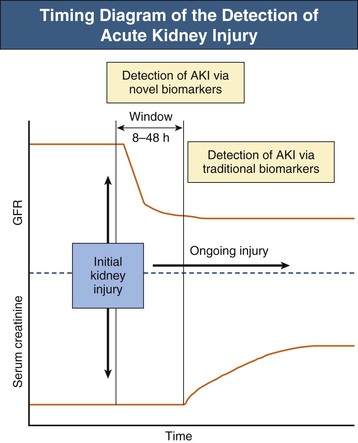
Figure 71-2 depicts the continuum of kidney injury severity and a number of biomarkers that have been proposed to be useful in early detection. Serum cystatin C, a cysteine protease inhibitor that is freely filtered at the glomerulus and normally reabsorbed by proximal tubule cells, may be more sensitive than serum creatinine concentrations at detecting small reductions in GFR.16 Urinary cystatin C has been shown to detect AKI in multiple clinical settings including after cardiac surgery, sepsis-AKI, and delayed graft function after kidney transplantion.15
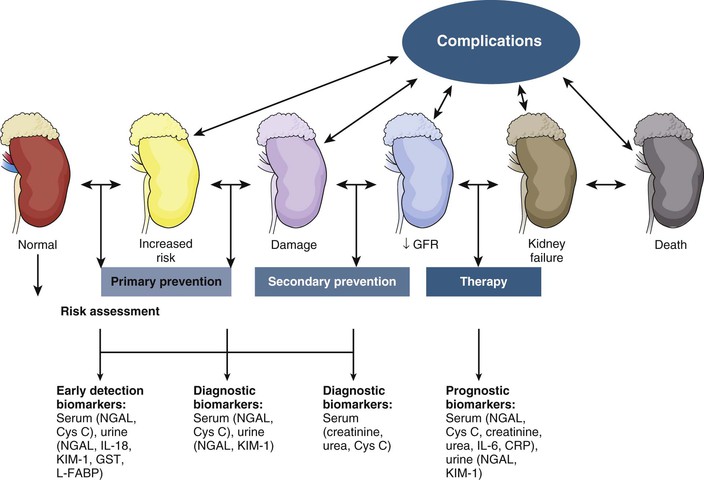
Kidney injury molecule 1 is a cell membrane glycoprotein that is upregulated in proximal tubular cells injured by ischemia or nephrotoxins in animals and humans. KIM-1 can act as a nonmyeloid phosphatidylserine receptor that transforms epithelial cells into semiprofessional phagocytes.17 The ectodomain of this membrane-associated mucin-rich molecule is shed into the urine of human and rodent kidneys with renal injury, but it is not found in urine produced by healthy kidneys. Urinary levels are increased specifically with AKI resulting from ischemia or toxin exposure.18,18a
Neutrophil gelatinase–associated lipocalin (NGAL), a protein expressed in proximal and distal renal tubule cells as well as neutrophils, binds and traffics free iron. It also mediates the tubular response to epidermal growth factor and is thereby involved in the progression of renal disease.19 Urinary NGAL levels are increased in the setting of tubular stress or injury, but not in pre-renal disease.20 NGAL is the most studied renal biomarker, with a large number of studies correlating urinary NGAL levels to the early detection of AKI.15
Interleukin (IL)-18 is an inflammatory cytokine found in macrophages and proximal tubule cells. Urinary IL-18 levels are upregulated in the setting of renal ischemic injury in multiple clinical settings: general ICU care, acute respiratory distress syndrome, contrast nephropathy, and cardiac surgery.15
Novel biomarkers of AKI have not reached clinical practice; however, they hold potential for detecting AKI early, identifying minor kidney injuries that do not raise serum creatinine concentrations, monitoring therapeutic benefits of novel treatment interventions, and specifying the cause of AKI. Whether the additional cost of screening for AKI or whether early detection of AKI will permit research that produces beneficial therapies for human AKI has not been established.20
Diagnostic Approach to Acute Kidney Injury
The basic diagnostic approach to patients with AKI is to determine the cause (see Chapter 69). This process should start by excluding or correcting both pre-renal and post-renal causes. In hospitalized patients, determining the correct causative diagnosis often involves selecting the most probable cause among many potential choices.21 In this setting, assessing urine volume can narrow the differential diagnosis, dividing AKI into oliguric (less than 500 ml of urine output per day) and nonoliguric causes.
To correctly identify the cause of AKI, one needs an understanding of the natural history of AKI from different causes, a chronologic sequence of events preceding the AKI, and analysis of the available patient data. Although the differential diagnosis for AKI in hospitalized patients is large, a careful history and physical examination and basic laboratory tests often suffice for diagnosis (Table 71-3).21
Table 71-3
Differential diagnosis by pathophysiologic classification of AKI.
ACE, Angiotensin-converting enzyme; ARBs, angiotensin receptor blockers; GI, gastrointestinal; HUS, hemolytic uremic syndrome; NSAIDs, nonsteroidal anti-inflammatory drugs; PPIs, proton-pump inhibitors; RPGN, rapidly progressive glomerulonephritis; SLE, systemic lupus erythematosus; TTP, thrombotic thrombocytopenic purpura.
| Differential Diagnosis by Pathophysiologic Classification of Acute Kidney Injury (AKI) | |
| Cause | Comments |
| Pre-renal | 30%-60% of AKI |
| Volume depletion | Renal losses, GI losses, hemorrhage |
| Decreased cardiac output | Right- or left-sided heart failure, cardiac tamponade |
| Systemic vasodilation | Sepsis, anaphylaxis, anesthetics |
| Afferent arteriolar vasoconstriction | NSAIDs, calcineurin inhibitors, radiocontrast, hepatorenal syndrome, hypercalcemia |
| Efferent arteriolar vasodilation | ACE inhibitors, ARBs |
| Intrinsic | Approximately 40% of AKI |
| Acute tubular injury | |
| Ischemic | |
| Nephrotoxic (drug) | Aminoglycosides, lithium, amphotericin, pentamidine, cisplatin, ifosfamide, radiocontrast |
| Nephrotoxic (pigment) | Rhabdomyolysis, intravascular hemolysis |
| Acute interstitial nephritis (AIN) | |
| Drug induced | Penicillins, cephalosporins, NSAIDs, PPIs, allopurinol, rifampin, sulfonamides |
| Infection related | Pyelonephritis, viral nephritides |
| Autoimmune diseases | Sjögren syndrome, sarcoidosis, SLE |
| Malignancy | Lymphoma, leukemia |
| Intratubular obstruction | |
| Paraprotein | Immunoglobulin light chains |
| Crystals | Acute phosphate nephropathy, tumor lysis syndrome, ethylene glycol, acyclovir, indinavir, methotrexate |
| Acute glomerulonephritis | Postinfectious, cryoglobulinemia, RPGN, SLE |
| Macrovascular | Increased renal vein pressure from increased intra-abdominal pressure, bilateral renal vein thrombosis, bilateral renal artery emboli |
| Microvascular | Atheroembolic disease, HUS, TTP, scleroderma renal crisis, malignant hypertension |
| Post-renal (Obstruction) | Approximately 10% of AKI |
| Intrinsic | Bilateral ureteral stones, bladder outlet obstruction (prostatic enlargement or blood clot), neurogenic bladder |
| Extrinsic | Retroperitoneal fibrosis, metastatic cancer |
Acute Kidney Injury Versus Chronic Kidney Disease
On occasion, it can be difficult to determine if a patient with renal failure has AKI or AKI superimposed on CKD. The patient’s history and information about prior serum creatinine values are invaluable for differentiation of AKI and CKD. Ultrasonographic evidence of small, scarred kidneys is consistent with CKD. Notably, CKD caused by diabetic nephropathy, infiltrative disorders such as amyloidosis, human immunodeficiency virus–related nephropathy, or polycystic kidney disease can be present with normal or increased kidney size. The findings on presentation of normocytic anemia, hyperparathyroidism, peripheral neuropathy, and broad waxy casts in the urinary sediment would suggest CKD. Patients with CKD are at high risk for the development of episodes of AKI.22 For patients with AKI superimposed on CKD, knowledge of prior serum creatinine concentrations is required to determine the degree of potentially reversible AKI.
Clinical Assessment
The evaluation of a hospitalized patient with AKI should begin with a complete medical history and review of the hospital records. Once the prior serum creatinine concentration or other evidence of underlying kidney disease has established the level of baseline kidney disease, the history should be directed toward the events preceding the AKI. These events may be part of a systemic disease process (e.g., sepsis, rhabdomyolysis), an inpatient event (e.g., surgical procedure, radiocontrast exposure, nephrotoxic medication exposure), or an outpatient event (e.g., medication or drug use, intravascular volume contraction from diarrhea or vomiting). Particular attention should be paid to the medication record including the use of nonsteroidal anti-inflammatory drugs (NSAIDs), renin-angiotensin-aldosterone antagonists, and antibiotic medications. Other features of the history include the use of Chinese herbs that contain aristolochic acid, a known nephrotoxin, or the use of a synthetic cannabinoid, a newly identified potential nephrotoxin.23
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


