Essentials of Diagnosis
General Considerations
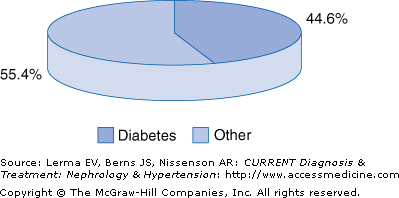
Diabetic nephropathy is a serious public health concern because it has become the leading cause of end-stage renal disease (ESRD) in most developed countries and is associated with increased cardiovascular mortality. Tracking both the incidence and prevalence of ESRD attributed to diabetes underscores its annual growth rate over the past decade in excess of 9%. According to the 2004 report of the U.S. Renal Data System (USRDS), in 2002, of 419,263 patients in the United States receiving either dialytic therapy or a kidney transplant, 149,614 had diabetes, a prevalence rate of 35.6%. The incidence rate was 44.5% in 2002, with 42,665 of 149,614 new (incident) cases of ESRD attributed to diabetes (Figure 54–1). This increase is mainly attributed to an increase in the occurrence of diabetes, especially type 2 diabetes; the extended life span of diabetic patients due to improved management of comorbid conditions; and the acceptance of patients for replacement therapy who in the past were excluded.

Diabetic nephropathy is characterized by an initial period of glomerular hyperfiltration associated with progressively increasing proteinuria, followed by a gradual decline in the GFR, eventually resulting in renal failure. Diabetic nephropathy afflicts 35–40% of type 1 and type 2 diabetic patients. While the natural history of diabetic nephropathy is well studied in type 1 diabetic patients, recent studies have shown a similar course of diabetic nephropathy in type 2 diabetic patients as well. In the past 2 decades, much has been learned about the possible pathogenesis of diabetic nephropathy leading to the development and use of specific therapies that have been effective in slowing progression to renal failure.
A cumulative incidence of diabetic nephropathy has been documented after 20–25 years of diabetes in both type 1 and type 2 individuals. Recent studies demonstrated that present treatment strategies substantially reduce the progression and incidence of diabetic nephropathy in type 1 diabetes. For example, a study from Sweden showed a substantial decline in albuminuria after 25 years of diabetes from 30% in patients in whom diabetes developed from 1961 to 1965 to 8.5% in those with onset from 1966 to 1970 and 13% in those diagnosed from 1971 to 1975. Similarly, the Steno Diabetes Center reported that in the same cohorts, the cumulative incidence of diabetic nephropathy after 20 years fell from 31.3% to 13.7%. Improved glycemic control, better control of blood pressure, and reduced prevalence of smoking were associated with the lower incidence of nephropathy.
In contrast to the decreasing incidence of diabetic nephropathy in type 1 diabetes, the incidence of diabetic nephropathy associated with type 2 diabetes mellitus has been increasing over the past 50 years, so that in the United States about 44% of all patients beginning ESRD replacement therapy have diabetes compared to 25–50% in Europe and about 25% in Australia. The incidence of diabetic nephropathy is about 1–2% per year in patients with type 1 diabetes.
Among young nonwhite patients with type 2 diabetes, such as Pima Indians, Japanese, and African-Americans, the incidence of nephropathy is similar to that of type 1 diabetes. However, the incidence of diabetic nephropathy is much lower in elderly white type 2 diabetic patients than in nonwhite patients.
Several large population-based studies found substantial racial and ethnic differences in the incidence rate of ESRD attributed to type 2 diabetes. The highest incidence of ESRD secondary to diabetes has been reported in Native Americans followed by Hispanics and African-Americans. In Pima Indians the cumulative incidence of ESRD after the onset of clinically detectable proteinuria is 40% at 10 years and 61% at 15 years. By contrast, ESRD developed in only 11% of white patients after 10 years of proteinuria and in 17% after 15 years. These ethnic and racial differences in the incidence of diabetic nephropathy reflect a complex and still poorly understood interplay between genetic and environmental factors.
Among patients starting renal replacement therapy, the incidence of diabetic nephropathy doubled from the years 1991 to 2001. Fortunately, the rate of increase is slowing down, probably because of adoption in clinical practice of several measures that contribute to the early diagnosis and delay of diabetic nephropathy, thereby slowing the progression to clinically noted renal disease. However, implementation of effective preemptive therapy in diabetic patients falls far below desirable goals.
Pathogenesis
The key to the development of diabetic nephropathy is hyperglycemia, which is postulated to mediate its effects in several different ways. First, glucose in high concentrations may be directly toxic to cells, altering cell growth and gene and protein expression, thus increasing extracellular matrix (ECM) and growth factor production. Second, hyperglycemia may induce its adverse effect indirectly through the formation of metabolic end products such as oxidative and glycation products. In vitro, transforming growth factor (TGF)-β modulates ECM production in glomerular mesangial and epithelial cells. In addition, TGF-β inhibits the synthesis of collagenases and stimulates production of metalloproteinase inhibitors, an effect that could lead to reduced degradation of ECM and hence ECM accumulation. High glucose concentrations also increase TGF-β mRNA expression in renal cells, stimulating TGF-β mRNA expression and bioactivity, cellular hypertrophy, and collagen transcription in proximal tubules, providing in vitro evidence for a role of TGF-β in the development of diabetic nephropathy.
Three of several pathways by which hyperglycemia may induce diabetic nephropathy are actively being explored.
In health, reducing sugars such as glucose react nonenzymatically and reversibly with free amino groups in proteins to form small amounts of stable Amadori products (eg, hemoglobin A1c) through Schiff base adducts. In normal aging, spontaneous further irreversible modification of proteins by glucose results in the formation of advanced glycation end-products (AGEs), a heterogeneous family of biologically and chemically reactive compounds with cross-linking properties. This process of protein modification is amplified by the high ambient glucose concentration present in diabetes. In cultured glomerular endothelial and mesangial cells in vitro, glycated albumin and AGE-rich proteins have been shown to enhance the expression of type IV collagen and TGF-β1 and increase protein kinase C (PKC) activity. When performed under physiological glucose conditions, in vitro studies provide evidence that early glycation products may contribute to the pathogenesis of diabetic glomerulopathy independently of glucose.
Aminoguanidine, a hydrazine-like compound, reacts with early glycation products inhibiting further AGE formation. Aminoguanidine retards the development of nephropathy and other complications of diabetes in long-term experimental rats.
Other AGE inhibitors and AGE cross-link breakers are able to ameliorate diabetic nephropathy in experimental animals.
The enzyme aldose reductase (AR) converts a variety of toxic aldehyde derivatives from lipid peroxidation to inactive alcohol.
AR is the rate-limiting enzyme in the polyol pathway, and facilitates the reduction of glucose to sorbitol. Sorbitol dehydrogenase converts sorbitol to fructose using nicotinamide adenine dinucleotide (NAD). In hyperglycemia, when the pathway of glucose to glucose-6-phosphate is saturated, excess glucose enters the polyol pathway and aldose reductase is activated, resulted in an accumulation of sorbitol. In in vitro experiments in mesangial cells, increased expression of glucose transporter 1 leads to increased AR expression and activity along with sorbitol accumulation and increased PKC-α protein levels, promoting stimulation of matrix protein synthesis. Numerous experimental and clinical studies with different AR inhibitors (ARI) implicate the diabetes-induced increased flux of glucose through the polyol pathway in the development of diabetic retinopathy and neuropathy; however, only a few studies have investigated the influence of ARI in diabetic nephropathy.
PKC is a family of serine-threonine kinases, consisting of at least 10 structurally related isoforms that regulate a variety of cell functions including proliferation, gene expression, cell differentiation, cell migration, and apoptosis. In vitro studies have shown that PKC is activated in vascular tissues and glomerular mesangial cells exposed to high glucose concentration. Activated PKC increases production of cytokines and ECM and vasoconstrictor endothelin-1. These changes contribute to basement membrane thickening, vascular occlusion, and increased permeability. Several PKC inhibitors used in experimental diabetes yielded promising results.
Ruboxistaurin (LY333531) mesylate, a bisindolymaleimide, shows a high degree of specificity within the protein kinase gene family for inhibiting PKC-β isoforms. In experimental rodent models of diabetes, ruboxistaurin normalized glomerular hyperfiltration, decreased urinary albumin excretion, and reduced glomerular TGF-β1 and ECM protein production, despite continued hypertension and hyperglycemia.
Clinical Findings
In health, daily urinary excretion of albumin is less than 25 mg. Diabetic nephropathy follows a characteristic course starting with microalbuminuria defined as albuminuria ranging from 30–299 mg/24 hours or 20–199 μg/minute to overt proteinuria defined as albuminuria ≥300 mg/24 hours or ≥200 μg/minute and worsening azotemia. There are several clinical similarities between diabetic nephropathy in patients with type 1 diabetes and in patients with type 2 diabetes; however, the clinical course differs in some respects between these two groups. In patients with type 1 diabetes in whom nephropathy develops the clinical course is relatively well defined. Nephropathy usually becomes clinically evident after 15–25 years of diabetes and almost always progresses to ESRD.
However, because of the frequently insidious onset of type 2 diabetes, the advanced age of many patients, and the common presence of coexisting vascular disease and hypertension, early renal involvement is frequently missed. In elderly patients with type 2 diabetes, it is not always clear whether renal failure is due solely to or even caused by diabetes. However, in young patients with type 2 diabetes, recent studies have shown a course similar to that in type 1 diabetes. Thus, the clinical course of diabetic nephropathy is best defined in type 1 diabetes and proceeds through several stages (Figure 54–2).
At the onset of type 1 diabetes, the GFR is above normal up to 140% in the majority of individuals. No single pathogenesis fully explains both the nephromegaly and glomerular hyperfiltration characteristic of type 1 diabetes; a correlation between renal enlargement and glomerular hyperfiltration has been inferred from the correction of both perturbations after the establishment of euglycemia. Intensive insulin therapy normalizes hyperglycemia and corrects glomerular hyperfiltration; the GFR begins to decline within 8 days of initiation of insulin therapy and falls further during 3 months of insulin treatment. A substantial subset of individuals (˜25–40%) with type 1 diabetes achieving usual levels of plasma glucose under insulin therapy continues to manifest a persistently elevated GFR; it is within this subgroup of hyperfiltering diabetic patients that the initial reductions in the GFR are first noted, with progression to clinical nephropathy.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


