, Omer A. Raheem1 and Ahmed Shabaik2
(1)
Department of Urology, University of California San Diego, San Diego, CA, USA
(2)
Department of Pathology, University of California, San Diego Medical Center, 200 West Arbor Drive, San Diego, CA 92103-8720, USA
Keywords
Renal cell carcinomaClear cellPapillaryChromophobeGradingStagingTreatmentIntroduction
With approximately 65,000 cases and 15,000 deaths, renal cell carcinoma (RCC) is the third most common and deadliest urologic malignancy, accounting for 2 % of solid malignancies diagnosed in the United States (USA), making it the sixth most common malignancy in men and the seventh most common malignancy in women [1, 2]. Despite a robust downward stage migration due to increasing utilization of radiological imaging modalities and diagnoses in incidental settings, deaths from RCC continue to rise, raising the scepter of an even higher death rate in the absence of the impact of incidentally discovered disease. The underlying reasons for this are unclear, but are likely related to as of yet ill-defined complex interaction between environmental risk factors and metabolic drivers (obesity, hypertension) which give rise to RCC and continue to drive incidence up even as other prominent risk factors may be on the decline (smoking/tobacco use), lack of overarching successful systemic therapeutic strategy, and an aging population [3–7].
This chapter will review the major histopathological categorization and subtypes of RCC and major benign cortical tumors, staging classifications, as well as therapeutic strategies and options for different stages and ongoing controversies regarding management.
Renal Tumor Pathology and Grading
Perhaps the earliest description of a renal tumor was that offered by Daniel Sennert in his textbook Practicae Medicinae in 1613. Under the term “Scirrhus renum,” Sennert stated, “Sometimes the kidneys are attacked by hard growths and hard swellings, which often happen following poorly cared for inflammation. Moreover the hard swelling of bad kidneys which has the capacity to throw a person into cachexia and dropsy, is for the greater part incurable.”
The first use of the word “Conventional” RCC was in the 1997 WHO classification of renal neoplasms, and it was used to denote the clear cell form of RCC. However, the editor’s selection as a title for this chapter “Conventional Forms of Renal Neoplasia” intends to include more than just that. For this reason in this chapter, we will try to discuss the most common forms of renal neoplasms known to pathologists for some time before the plethora of the recently recognized special forms of renal neoplasms that started around the mid-1980s with the introduction of chromophobe RCC by Theones and his colleagues. In this chapter, we will discuss clear cell RCC, multilocular cystic clear cell renal cell neoplasm of low malignant potential (multilocular cystic renal cell carcinoma), papillary RCC, papillary adenoma, chromophobe RCC, and oncocytoma .
Clear Cell Renal Cell Carcinoma
Clear cell renal cell carcinoma (CCRCC) is the most common renal malignancy . It has been initially named the Grawitz tumor in reference to Grawitz who in 1883 recognized microscopically the presence of adrenal rests in the renal cortex, gave it the name “struma suprarenalis aberrata” (Fig. 5.1 ). He postulated that they are precursors of renal neoplasms [8]. However, in 1893 Sudeck published his observation of finding atypical tubules adjacent to renal tumors, postulated that these are the origin of renal tumors, and challenged Grawitz theory [8]. Such finding has been recently supported by one publication [9]. Despite Sudeck’s challenge, in 1894 Lubarsch supported Grawitz theory and coined the term “hypernephroid tumor” which was later on modified to “hypernephroma” by Birch-Hirschfeld [8]. This term remained in use until the late 1970s.
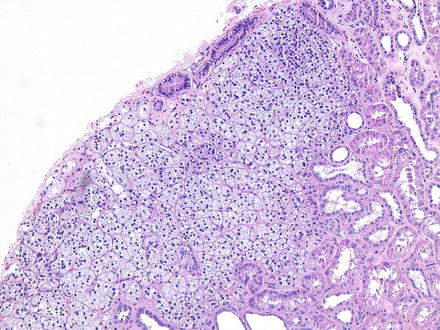
Fig. 5.1
Adrenal rest in renal cortex . 100× magnification
CCRCC occurs in sporadic and familial forms . Sporadic CCRCC is mostly found in the 6th decade with male to female ratio of 2–3:1 as solitary mass of the renal cortex commonly protruding on the renal surface as a globular or bosselated mass. Multiple or bilateral tumors or early age of onset should raise concern for the familial forms associated with von Hippel-Lindau (VHL) syndrome or constitutional chromosome 3 translocation. Familial cases comprise less than 5 % of all cases of CCRCC. A band of fibrosis of variable thickness (pseudocapsule) is present at the interface of the tumor with the nonneoplastic kidney. Generally, the cut surface is solid and golden yellow in color due to high fat content, but commonly displays foci of hemorrhage and necrosis and cyst formation. Calcification and even ossification may be encountered. The size is variable, but the majority of tumors nowadays are less than seven centimeters due to advances in the imaging and surgical techniques.
Microscopically as the name indicates, the tumor consists of cells with optically clear cytoplasm due to high content of lipid and glycogen, which dissolve during routine processing. A population of cells with eosinophilic cytoplasm can be found and rarely may be the predominant cell type (Fig. 5.2 ). The cell border is distinct but not prominent. The cells are arranged in solid sheets and nests or alveoli and acini that may dilate to form microcystic or macrocystic spaces or combination of patterns. These spaces may be filled with serous fluid or blood (“blood lakes”). An elaborate network of thin-walled blood vessels invests these structures. The nuclei are usually round and uniform with evenly distributed chromatin. The size of the nucleus and the prominence of the nucleolus are the basis for the Fuhrman grading system. A host response of tumor infiltrating lymphocytes is usually present but can be very variable in intensity. Rarely a granulomatous response can be seen around tumor cells (Fig. 5.3 ). Host response and tumor necrosis with subsequent organization may cause tumor regression and reduce the tumor to a small scar.
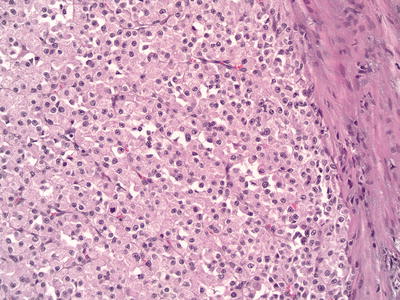
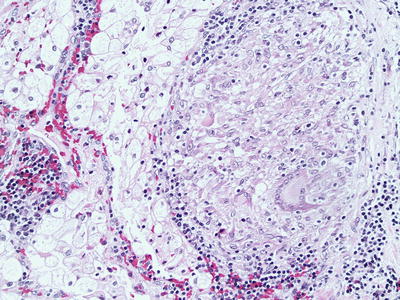
Fig. 5.2
Clear cell renal cell carcinoma with predominantly eosinophilic cells . 200× magnification
Fig. 5.3
Granulomatous host response to clear cell renal cell carcinoma. 200× magnification
Immunohistochemistry can be helpful with the diagnosing CCRCC particularly in limited samples like core biopsies and fine needle aspiration. CCRCC is usually positive with the RCC marker, vimentin, PAX2, PAX8, CD10, carbonic anhydrase IX, and CK18 and usually negative with CK5, CK6, CK7, CK20, HMWKs, and CD117 [9]. AMACR and E-cadherin can be variable [10].
The tumor grade is second to the tumor stage in predicting prognosis. The most widely used grading system is the nuclear grading system published by Fuhrman et al. in 1982 [11]. It is a four-tier grading system based on the size of the nucleus and the prominence of the nucleolus at the 10× objective. Grade 1 tumors have nuclei that are less than 10 μm with dense chromatin and invisible nucleoli at the 100× magnification or higher. Grade 2 nuclei are less than 15 μm with finely granular chromatin and invisible nucleoli at 100× magnification but may be visible at 200×. Grade 3 nuclei are less than 20 μm and nucleoli are easily visible at 100× magnification. Grade 4 tumors display nuclei that are larger than 20 μm with noticeable hyperchromasia and pleomorphism and contain single or multiple macronucleoli. CCRCC can show areas with variable grades; however, the tumor grade is assigned based on the highest grade encountered. In 2013, ISUP proposed a modification of the Fuhrman grading system, which relies solely on the prominence of the nucleoli for grades 1–3. ISUP grade 4 tumors should encompass tumors with rhabdoid or sarcomatoid differentiation or those containing tumor giant cells or showing extreme nuclear pleomorphism with clumping of chromatin [12].
Staging of CCRCC is not discussed here extensively; however, CCRCC commonly spreads via blood stream. About 75 % of metastatic CCRCC are seen in the lungs. It is also known to metastasize to unusual sites like the eye, thyroid, parotid glands, pancreas, and cerebellum even after many years, which can reach up to 30 years. In such odd sites, the metastatic CCRCC may mimic primary tumors of these organs (e.g., clear cell myoepithelioma of parotid or hemangioblastoma of the central nervous system, which also shares the VHL mutation with CCRCC) [13]. In such situations, one may miss the diagnosis particularly if the history of prior RCC is not available due to long interval or due to patient changing hospitals. The clear cell morphology and the intricate delicate vascular network should stimulate the alert pathologist to think about possible metastatic CCRCC. The role of immunohistochemistry in directing the workup of such situations cannot be ignored.
Although most CCRCC are sporadic, cytogenetic studies have detected chromosomal abnormalities in the majority of them including 3p deletions involving the VHL locus at 3p25–26 [14, 15], the familial human RCC chromosomal translocation point at 3p13–14 [16], and 3p21–22 [17]. Chromosome 3p deletions were detected in very small CCRCC suggesting it to be an early event in its development [18]. Other chromosomal abnormalities like 9p loss [19] and 14q deletions [20] have been found in patients with higher grade and stage and they correlate with poor outcome.
Multilocular Cystic Clear Cell Renal Cell Neoplasm of Low Malignant Potential (Multilocular Cystic Renal Cell Carcinoma )
A tumor closely related to CCRCC, these in its pure form have never been reported to have progressed, recurred, or metastasized and are seen in adults and more commonly in males. Previously regarded as a “carcinoma,” in 2013 ISUP adopted a change in the nomenclature for these tumors to reflect its low malignant potential [21]. They are well-circumscribed masses with fibrous capsule and made up entirely of cysts. The cysts are lined by clear cells with grade 1 nuclei and the septa between the cysts may contain small non-expansile groups of clear cells with grade 1 nuclei [22, 23].
Papillary Renal Cell Carcinoma
Papillary renal cell carcinoma (PRCC) is the second most common malignant renal neoplasm and comprises about 10 % of RCC. Its age and sex distribution and clinical presentation are similar to that of CCRCC [24, 25]. Angiography studies suggest relative hypovascularity of PRCC [25]. Grossly PRCC is more prone to show multifocality and bilaterality particularly in cases of hereditary PRCC [26]. They frequently show areas of hemorrhage, necrosis, and cystic change. Well-circumscribed tumors have fibrous capsule. In the 1986 Mainz classification of renal tumors, PRCC was named chromophil RCC in contrast to the chromophobe RCC, and this was mainly due to its microscopic composition of either basophilic or eosinophilic cells or combination. This chromophilic classification did not convey much prognostic significance. More recently Delahunt and Eble [27] proposed reclassifying PRCC into type 1 and type 2 based on that in type 1 the papillary fronds are lined by a single layer of small cells with scant cytoplasm (Fig. 5.4 ), while in type 2 tumor papillae are lined by larger cells with eosinophilic cytoplasm with pseudostratified nuclei of usually higher grade (Fig. 5.5 ). This classification proved to be of better prognostic significance [27, 28]. Type 1 is commonly multifocal, while type 2 is unifocal. PRCC is not always made entirely of papillary structures but can show variable proportions of papillae and tubules. Large cystic areas with papillary fronds can also be found. Aggregates of foamy histiocytes and hemosiderin-laden macrophages can been seen mostly in the papillary cores. The lining epithelium may show hemosiderin granules decorating its luminal border. Calcifications and psammoma bodies can be found in PRCC. The so-called solid variant of PRCC is due to predominance of tubular and glomeruloid growths or to very compact papillae [29]. Sarcomatoid foci can be seen in up to 5 % of type 1 and type 2 PRCC, mainly as high-grade spindle cells [27].
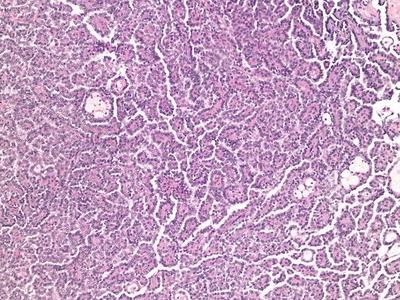
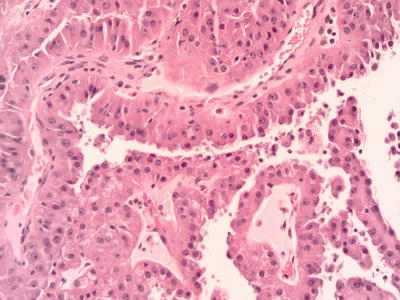
Fig. 5.4
Type I papillary renal cell carcinoma. 100× magnification
Fig. 5.5
Type II papillary renal cell carcinoma. 400× magnification
By immunohistochemistry , PRCC is usually positive for RCC marker, CK7, vimentin, AMACR, PAX2, PAX8, and CD10 and usually negative for HMWK, E-cadherin, and CD117 and negative for uroplakin and p63, which help differentiate from papillary urothelial carcinoma if it is in the differential diagnosis of a papillary renal neoplasm. TFE3 is also negative in PRCC, which will be helpful in differentiating it from an Xp11 translocation carcinoma with papillary architecture [10, 30]. The non-sporadic (familial) PRCC are seen in patients with the hereditary PRCC syndrome and Birt–Hogg–Dube syndrome and these are usually type 1 [26, 31], while type 2 is usually seen in patients with the hereditary leiomyomatosis and RCC syndrome [32, 33].
The commonest cytogenetic abnormalities in PRCC are trisomy of chromosomes 7 and 17 and loss of chromosome Y [34]. These karyotypic abnormalities are diagnostic of PRCC even if the tumor lacks the papillary architecture. In their absence, a tumor cannot be classified as PRCC even if it displays papillary architecture. The type of PRCC, nuclear grade, and stage correlate with prognosis and survival [28]. However, in multivariate analysis, only the tumor stage seems to be the only significant variable [35].
Papillary Adenoma
The term papillary adenoma is restricted to tumors that are 0.5 cm or less in diameter and composed of papillary or tubulo-papillary structures. With this definition they are the most common epithelial neoplasm of the kidney as suggested by autopsy studies which show exponential increase in their incidence from 10 % in patients younger than 40 years to 40 % in patients older than 70 years [36, 37]. They are also seen with higher frequency in kidneys of patients on long-term dialysis regardless of age [38, 39]. Grossly, they are frequently encountered as subcapsular yellow or greyish white nodules. Microscopically they are similar to type 1 PRCC (Fig. 5.6 ) but occasionally can look like type 2 also. Most tumors are not encapsulated, but if a capsule is present, it is usually thin. Foamy macrophages and psammoma bodies are frequently present. Papillary adenoma shares the same genetic abnormality like PRCC with loss of Y and trisomy 7 and 17.
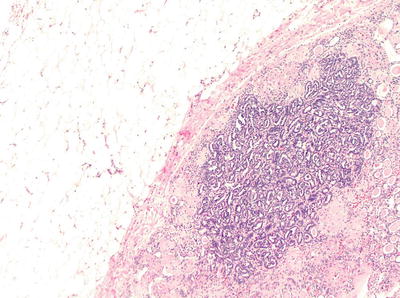
Fig. 5.6
Papillary adenoma . 100× magnification
Chromophobe Renal Cell Carcinoma
Theones described chromophobe renal cell carcinoma (CRCC) in 1985 as renal carcinoma with large pale cells with prominent cell membrane [40]. It accounts for about 5 % of all renal tumors mostly encountered in the 6th decade with equal incidence in men and women [41]. Sporadic and hereditary forms exist.
Grossly they are usually solid circumscribed tumors with variegated light brown to tan cut surface. Microscopically they are characterized by large polygonal cells with clear to pale eosinophilic finely reticulated cytoplasm with prominent cell membrane. The nuclei are usually wrinkled with small nucleoli and a perinuclear halo (Fig. 5.7 ). Binucleation is common. An eosinophilic variant of CRCC has been described in which the cell cytoplasm is eosinophilic without reticulation [42]. Sarcomatoid areas can rarely be encountered in CRCC [43–46]. The cytoplasm of the CRCC cells stains positive with Hale’s colloidal iron stain [40, 47] (Fig. 5.8 ). Ultrastructurally, CRCC shows abundant microvesicles and the eosinophilic type is rich in mitochondria. Grading of CRCC is controversial as many well-behaving and low-stage tumors show innate atypical nuclei [48]. The Fuhrman grading system was shown to be not applicable for CRCC [49, 50]. An alternative histologic grading system was proposed, but further validation is needed [50, 51].
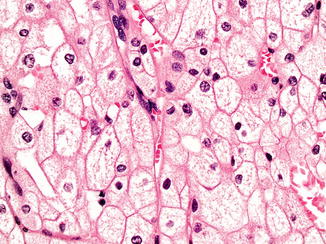
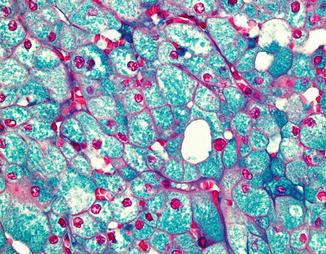
Fig. 5.7
Chromophobe renal cell carcinoma, classic variant. 400× magnification
Fig. 5.8
Positive colloidal iron stain in chromophobe renal cell carcinoma. 400× magnification
The immunohistochemical profile of CRCC is positive for CK7 (Fig. 5.9 ), CK18, E-cadherin, kidney-specific cadherin, PAX8, and CD117 and negative for vimentin, AMACR, and variable with RCC marker and CD10, but mainly negative [10, 30, 47]. CRCC shows numerous chromosomal loss, which usually leads to hypodiploidy on DNA analysis [52, 53]. CRCC has good prognosis with no mortality at 5 years and 10 % mortality at 10 years [54]; however, sarcomatoid transformation is a poor prognostic indicator [45].
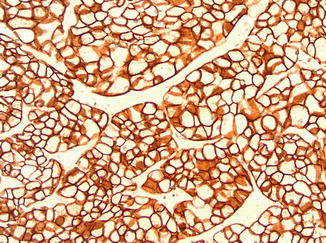
Fig. 5.9
Cytokeratin 7 in chromophobe renal cell carcinoma. 200× magnification
Oncocytoma
A benign renal neoplasm made up of oncocytic cells with abundant granular eosinophilic cytoplasm rich in mitochondria. It was first described by Klein and Valensi in 1976 [55]. It constitutes about 5 % of all renal epithelial neoplasms and it is most frequent in the 7th decade with a male to female incidence ratio of 2:1. Most tumors are sporadic. It is believed that both oncocytoma and chromophobe RCC share origin from the intercalated cells of the cortical collecting ducts [47, 52, 55–59].
Grossly, oncocytomas are discrete well circumscribed but not encapsulated tumors. The cut surface is “mahogany brown” solid with a stellate scar in about 40 % of cases. Punctate hemorrhage may be encountered but no gross evidence of necrosis. Microscopically the tumor is composed of oncocytes that are round to polygonal with deeply eosinophilic granular cytoplasm with round regular centrally located nucleus with prominent nucleolus. The oncocytes are arranged in compact groups of small solid nests, acini, tubules, microcysts, or a combination of these structures embedded in loose edematous or hypocellular hyalinized stroma. Occasional clusters may display atypical degenerative nuclei, which does not affect the biologic behavior of the tumor. Mitoses are rare to absent and if present they are typical mitoses. Abundant clear cells or true papillary architecture is not a feature of oncocytoma and if encountered a diagnosis other than oncocytoma should be sought. Rarely oncocytoma may extend into perinephric fat or be seen within vessels [58–60]. Such features do not affect prognosis. Being a benign tumor, oncocytoma should not be graded or staged. Oncocytomas do not metastasize, although one report has claimed that two of seventy cases had metastasis. One case metastasized to the liver, which was histologically confirmed, and the second case metastasized to the liver and bone, but this was not histologically confirmed and could have been metastasized from other tumors [59].
Oncocytoma being related to CRCC should be differentiated from it. Oncocytomas have no fibrous capsule, show no prominent cell membranes, and do not show diffuse reticular positive reaction with Hale’s colloidal iron stain, and nuclei are not wrinkled. Oncocytomas show overlapping immunophenotypic features with CRCC including the positivity for CD117. CK7 is usually negative in oncocytoma and if positive will be in the form of scattered positive cells (Fig. 5.10 ), in contrast to CRCC, which shows diffuse positive reaction. Few studies reported loss of chromosome Y and 1 in oncocytoma [61, 62].
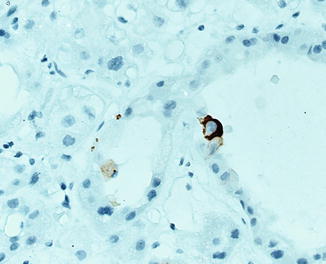
Fig. 5.10
Cytokeratin 7 in oncocytoma
Staging of Renal Cell Carcinoma
Staging of RCC is based on the AJCC/TNM classification with its most recent iteration being in 2010, incorporating modifications based on increased understanding of tumor biology. The major change involved reclassification of T3a tumors with noncontiguous adrenal involvement (which had a worse prognosis to T4), while proposed erasure of the breakpoint between T1a and T1b at 4 cm was rejected, based on data suggesting that size appears to have continual prognostic significance, especially in the range of 2–6 cm [5, 6]. Nonetheless, the broad categories and breakdown of staging have remained unchanged and are as follows: stage I-T1N0M0 , with T1 tumors being defined as <7 cm maximum dimension, and the breakpoint between T1a and T1b being 4 cm; stage II-T2N0M0, with T2 tumors being defined as >7 cm and the breakpoint between T2a and T2b being 10 cm; stage III-T3N0/1M0, with T3a being revised to include tumors breaking the renal capsule and tumor thrombus being confined to the renal vein, T3b including tumors with thrombus into the subdiaphragmatic inferior vena cava (IVC), and T3c comprising tumor thrombus into the supradiaphragmatic IVC; and stage IV comprising T4 (with extension into adjacent organs) with AnyN/AnyM or TAny/withN (>1 regional lymph node metastasis) or M1 [63].
Evaluation and Workup of Cortical Renal Tumors
The cornerstone of evaluation is the history and physical . Careful attention should be paid to concurrent symptoms (e.g., hematuria or flank pain), as well as family history or symptomatic presentation which may guide further studies. Presence of physical exam findings may also indicate locally advanced (e.g., left-sided varicocele, lower extremity edema) or metastatic disease. The standard laboratory evaluation should include determination of renal function (BUN/creatinine and calculation of eGFR), liver function tests (including alkaline phosphatase), and a complete blood count. While several putative markers [nonspecific markers of systemic inflammation such as C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and platelet count] and tissue-based markers such as carbonic anhydrase IX have been demonstrated to have prognostic predictive utility, currently no single test or set of tests has gained widespread acceptance as an overarching tumor marker [3, 4].
Utilization of cross-sectional contrast-enhanced imaging studies (MRI or CT) remains as the confirmatory modalities of choice to assess local extent of disease and regional and abdominal metastases [64]. While data exist which demonstrate differential enhancement patterns between different tumor histologies, currently such data are of limited utility in clinical decision making [65, 66]. MRI may have added advantages in delineation of venous thrombus extent and allows utilization of contrast in patients with eGFR < 60; utilization of gadolinium contrast in patients with eGFR < 30 (severe CKD) is contraindicated given risk regarding progressive systemic fibrosis [67]. Chest imaging should be obtained as part of the workup, given the fact that the chest is the most common site of metastasis in RCC. Generally for lower-risk tumors, a chest X-ray is sufficient, with CT of the chest being preferred for higher-risk primary tumors or in patients with suggestive symptomatology. Current data do not support utilization of PET/CT for staging or follow-up of RCC, and utilization of adjunctive staging imaging (bone scintigraphy) or head CT should be limited to patients with elevated alkaline phosphatase (bone scintigraphy) or symptomatology (bone or neurological imaging) [6].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


