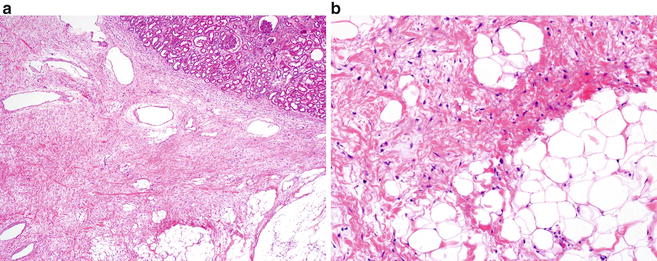
Fig. 10.1
Classic angiomyolipoma with various amounts of adipose tissue , smooth muscle, and thickened vessels (Hematoxylin eosin, 20×)
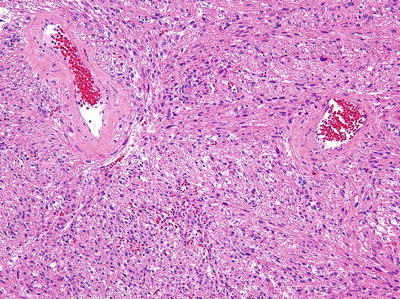
Fig. 10.2
Spindle cell variant of angiomyolipoma with a predominant smooth muscle component without significant fat (Hematoxylin eosin, 20×)
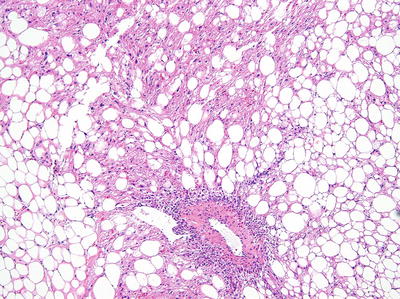
Fig. 10.3
Predominantly lipomatous angiomyolipoma (Hematoxylin eosin, 10×)
Immunohistochemically, AMLs express melanocytic markers (HMB-45, Melan-A/MART-1, microphthalmia transcription factor, and tyrosinase), smooth muscle markers (smooth muscle actin, h-caldesmon), and cathepsin K and are typically negative for keratins or other epithelial markers and PAX8. This immunoprofile is very useful in the distinction of AML from other differential diagnostic considerations.
Classic AML is associated with a benign outcome. Treatment consists primarily of surgical resection, although percutaneous renal biopsy can be used to establish a diagnosis and potentially obviate the need for surgery.
Epithelioid Angiomyolipoma
Epithelioid angiomyolipoma is identified on histological review and is characterized by the presence of plump epithelioid stromal cells. Two major morphologic patterns , which may occur pure or in combination, have been described: (a) carcinoma-like growth, characterized by large cells arranged in cohesive nests, alveoli, and sheets with intervening thin fibrovascular septa, and (b) epithelioid and plump spindle cells in diffuse growth [14] (Fig. 10.4). Compared to tumors with predominant spindle histology, epithelioid AML tends to show less smooth muscle marker expression with more widespread melanocytic marker expression as well as estrogen receptor expression [15].
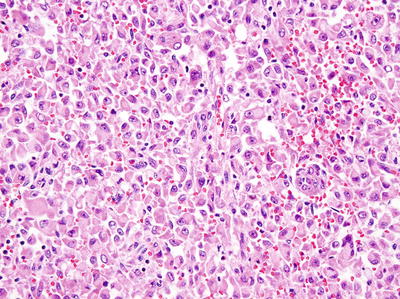
Fig. 10.4
Epithelioid angiomyolipoma with diffuse sheets of discohesive, plump, epithelioid cells (Hematoxylin eosin, 20×)
In contrast to classic AML, a series of 34 cases of epithelioid AML with available follow-up showed malignant behavior in 26 % of cases [16]. Another study of 31 cases showed recurrence or metastasis in 17 and 49 % of patients, respectively [14]. Larger tumor size, invasion outside the kidney, a carcinoma-like growth pattern, significant/diffuse nuclear atypia, and necrosis were among the factors associated with disease progression [14, 16].
Leiomyoma
Leiomyomas may arise from the renal capsule (most common), from muscularis of the renal pelvis, or from cortical vascular smooth muscle [17–19]. Most patients are adults and can present with pain or an abdominal/flank mass. Tumor size is variable, with one series reporting an average of 12 cm [18], while the morphology is quite bland with sparse mitotic activity. Calcification and cystic change have been described, but necrosis should not be present. As expected, leiomyomas express smooth muscle markers, but not melanocytic markers. The latter is of value in the distinction from the more common lipid-poor AMLs.
Hemangioma
Hemangiomas occasionally arise in the kidney and appear as capillary [20, 21] or cavernous blood-filled vascular spaces lined by a single layer of endothelial cells. Although certain hemangiomas can exhibit an infiltrative growth pattern, they lack the mitosis and nuclear pleomorphism seen in angiosarcoma.
Solitary Fibrous Tumor
Frequently arising in extrathoracic sites, this tumor can occasionally arise in the kidney. It characteristically exhibits fibroblastic differentiation with a prominent staghorn-like branching vascular pattern and areas of collagen deposition. In contrast to solitary fibrous tumors elsewhere that can be malignant, almost all of those arising in the kidney have reportedly behaved in a benign fashion [22, 23].
Juxtaglomerular Cell Tumor (JG Tumor)
This is a benign neoplasm that often affects adolescents and young adults. JG tumors secrete renin, and as such this entity may be suspected in patients with severe poorly controlled hypertension and marked hypokalemia. Headaches and polyuria have also been reported. These tumors are typically 5 cm or less in size and are usually solitary and well circumscribed. Histologically, JG tumors are composed of sheets of polygonal or spindled tumor cells with central round regular nuclei with a complex vascular hemangiopericytic pattern that entraps normal renal tubules [3]. These tumors express renin, vimentin, CD34, CD117, and α-smooth muscle actin. Recurrence or metastasis has not been reported, although these tumors may be lethal if left untreated. Surgical excision is curative for JG tumors.
Renomedullary Interstitial Cell Tumor
This is a common benign finding in adult autopsies and may be found incidentally in surgical resection specimens in patients >50 years of age [24]. This lesion has also been termed “medullary fibroma.” Most patients are asymptomatic, although rare cases have been associated with hematuria and urinary tract infections. These lesions arise from the interstitial cells of the medulla and, as such, are typically immunoreactive for smooth muscle actin, COX-2, and PGE2. Microscopically, the tumor is composed of small stellate or polygonal cells in a background of loose faintly basophilic stroma reminiscent of that seen in renal medulla. At the periphery, renal medullary tubules often are entrapped in the matrix [3].
Malignant Mesenchymal Neoplasms Involving the Kidney
Because of their usually larger size and invasive nature, malignant non-epithelial neoplasms are more frequently symptomatic with pain, a palpable mass, or hematuria being common presenting symptoms. On imaging, renal sarcomas are usually large, invasive tumors with areas of enhancement, necrosis, and occasionally calcifications [25].
Renal sarcomas are often large and locally invasive; therefore, radical nephrectomy is typically the primary treatment modality [25, 26]. The role of regional lymphadenectomy is widely debated for epithelial renal neoplasms, and there is no consensus regarding its use in malignant non-epithelial renal tumors. However, given that sarcomas may present with locoregional lymphadenopathy, a retroperitoneal lymphadenectomy may be of diagnostic and potentially therapeutic benefit. Complete surgical resection with no residual disease offers the best chance at cure [26]. These tumors may also be locally invasive, thereby requiring concomitant partial, or complete, resection of the involved structures (bowel, spleen, liver, psoas muscle, quadratus, ribs, vertebrae). Often the surgical team is comprised of multiple disciplines (urology, orthopedics, vascular and plastic surgery) and preoperative consultation and planning between surgeons is essential.
Malignant non-epithelial renal neoplasms are generally aggressive tumors with a poor prognosis. For renal sarcomas, the overall survival (OS) at 1, 3, and 5 years was 86.3, 40.7, and 14.5 %, respectively, according to one of the largest case series [26]. Other series report a relatively higher 5-year OS of 25 % [27]. Variables that appear to predict OS include complete surgical resection, tumor stage, and tumor grade [26, 27].
Leiomyosarcoma
This is the most common renal sarcoma accounting for 50–60 % of cases [28, 29]. It may arise from the renal capsule, the renal parenchyma, smooth muscle fibers of the renal pelvis, or the main renal vein [6]. Histologically, it is composed of eosinophilic spindle cells with distinct smooth muscle features arranged in a fascicular, plexiform, or haphazard growth pattern (Fig. 10.5). The presence of necrosis, nuclear pleomorphism, and more than a rare mitotic figure helps in differentiating leiomyosarcomas from leiomyomas.
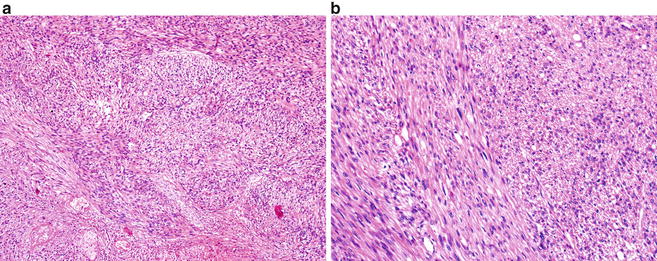
Fig. 10.5
Leiomyosarcoma , exhibiting spindle cells with distinct smooth muscle features arranged in a fascicular or haphazard growth pattern. (a) Hematoxylin eosin, 4×. (b) Hematoxylin eosin, 20×
In the setting of unresectable leiomyosarcomas or for palliation, chemotherapy and radiation have been employed. Chemotherapy regimen includes cisplatin, adriamycin, etoposide, ifosfamide, and vincristine. Radiotherapy median dose is typically around 42Gy (range 30–60) [26, 30]. Renal lymphomas are managed with chemotherapy, and a common regimen is CHOP (cyclophosphamide, adriamycin, vincristine, prednisone) [31].
Synovial Sarcoma
A characteristic feature of this mesenchymal neoplasm is the variable degree of epithelial differentiation that could be present. Histologically, synovial sarcoma displays monomorphic plump spindle cells with indistinct cell borders growing in short, intersecting fascicles or in solid sheets, often with prominent mitotic activity. At the molecular level, the tumor is characterized by a specific translocation t(X;18)(p11.2;q11). In contrast to soft tissue synovial sarcoma where the SYT-SSX1 gene fusion is more common than the alternative SYT-SSX2 form [32], the majority of renal synovial sarcomas have so far predominantly demonstrated the SYT-SSX2 gene fusion [33–35].
Liposarcoma
Although retroperitoneal liposarcomas commonly occur in the perirenal area, primary liposarcomas of the kidney are very rare. Histologically, well-differentiated liposarcoma is composed of a relatively mature adipocytic proliferation, frequently with overt variation in cell size, focal cytologic atypia (Fig. 10.6), and consistent MDM2 (12q15) amplification [36]. This tumor is often associated with multiple recurrences, with about 10 % undergoing dedifferentiation. The latter most frequently resembles undifferentiated pleomorphic sarcoma or high-grade myxofibrosarcoma. Approximately 40 % of dedifferentiated liposarcomas recur locally and 15–20 % metastasize [36].