John O. Connolly, Guy H. Neild
Congenital Anomalies of the Kidney and Urinary Tract
Congenital anomalies of the kidney and urinary tract can result in renal problems and renal failure. Almost half of children who develop end-stage renal disease (ESRD) have asymmetric, irregularly shaped kidneys.1,2 This appearance, often referred to as “bilateral renal scarring,” is frequently associated with lower urinary tract anomalies that include vesicoureteral reflux (VUR). The most serious conditions involve bladder outflow obstruction, and many anomalies are now detected antenatally.
These cases were previously described as reflux nephropathy or chronic pyelonephritis. With advances in genetics and developmental biology, however, it is becoming clear that many anomalies are caused by primary renal malformations, or renal dysplasia, often associated with congenital malformations of the ureter, bladder, and urethra. This is a change from the view that renal scarring and damage are secondary to the outflow problem and ureteral reflux. Although still somewhat debatable, the British Association for Pediatric Nephrology has stated that the clinical distinction between reflux nephropathy and renal dysplasia is arbitrary and unnecessary.1
Clinical Principles
Congenital renal tract abnormalities may present in one of five settings:
1. Antenatal diagnosis by fetal ultrasound screening
2. Failure to thrive in an infant or young child
3. Investigation of urinary tract infection (UTI)
4. An incidental finding in a child or adult
5. An adult with abnormal urinalysis, stones, hypertension, or renal impairment
The identification of these problems always poses the following questions:
Such patients fall into two broad groups. The first group of patients appear to have normal bladders without outflow obstruction and normal-caliber ureters when not micturating, described as having either primary VUR or primary renal dysplasia. The second group has some form of bladder outflow dysfunction that causes secondary VUR and dilated upper urinary tracts, the most common cause of which is posterior urethral valves in males.
As predicted by the Brenner hypothesis,3 small asymmetric kidneys with reduced glomerular filtration rate (GFR) develop all the features of glomerular hyperfiltration, with the onset of progressive renal failure signaled by increasing proteinuria. This can now be significantly modified by treatment with renin-angiotensin blockade.4,5 The details of antenatal and pediatric management of these patients are beyond the scope of this chapter, which focuses on management in adolescence and adult life.
Development of the Urinary Tract
The urinary tract develops from the cloaca and intermediate mesoderm in parallel with the early differentiation of the metanephric blastema (future kidney)6–8 (Fig. 52-1). At the fifth week of gestation, the mesonephric (wolffian) duct connects to the allantois and the cloaca. By the sixth week, the urorectal fold appears and divides this cavity, which separates the future urinary system (urogenital sinus) from the rectum (hind gut). Growth of the anterior abdominal wall between the allantois and the urogenital membrane is accompanied by an increase in size and capacity of this bladder precursor. The allantois remains attached to the apex of the fetal bladder and extends into the umbilical root, although it loses its patency and persists as the urachal remnant, the median umbilical ligament, which connects the bladder to the umbilicus. By the seventh week, there is a separate opening of the mesonephric duct into the bladder at what will become the vesicoureteral opening and the area known as the trigone. The distal part of the primitive urogenital sinus will form the definitive urogenital sinus. In females, this gives rise to the entire urethra and the vestibule of the vagina. In males, it gives rise to the posterior urethra, whereas the anterior urethra is formed from the closure of the urethral folds.
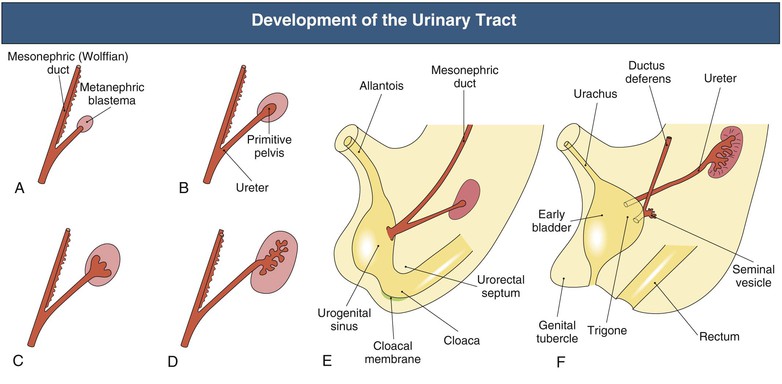
In the 6-week embryo, the mesonephric and paramesonephric (müllerian) ducts run in parallel. By 7 weeks, in the male, the müllerian duct starts to regress, and the wolffian duct will eventually develop into the epididymis and the caudal part of the vas deferens. In the female, the müllerian ducts fuse to become the uterovaginal cord, which opens into the urogenital sinus and will develop into the vagina.
As the urogenital tract develops, there is simultaneous development of the fetal kidney (Fig. 52-1). The ureteric bud arises from the distal end of the wolffian duct as an unbranched diverticulum (day 28) and invades the adjacent metanephric mesenchyme, initiating the branching collecting duct system within the primitive kidney. In the absence of the ureteral bud, the metanephric kidney does not form. Fetal urine reaches the bladder by 8 to 10 weeks and makes a significant contribution to the amniotic fluid by 16 weeks. Nephron formation is complete by 34 weeks’ gestation.
Renal development is orchestrated by the expression of transcription factors, growth and survival factors, and adhesion molecules.6,8,9 Mutations of genes encoding all classes of these molecules cause urinary tract malformations in mice.8–10 One family of transcription factor proteins contains the paired DNA-binding domain and is encoded by the PAX genes. Studies in mice show that these PAX genes regulate the development of brain, eyes, lymphoid system, musculature, neural crest, and vertebrae.11,12 PAX2 is expressed in the metanephros and in cell lineages that are forming nephrons and also in those that are destined to differentiate into the ureter, renal pelvis, and branching collecting duct system. The ablation of a single PAX2 allele in knockout mice causes impaired metanephric growth and fewer nephrons than normal as well as megaureter, a finding consistent with gross VUR.12 WT1, which is mutated in a proportion of Wilms tumors, is another transcription factor whose mutation is associated with abnormal urinary tract development.
Normal development appears to depend on urine flow from the fetal kidney. This requires peristalsis and no anatomic obstruction to flow. A number of diverse defects can now be unified as it becomes clear they play some key role in peristalsis. This may be caused by defects in smooth muscle development or innervations in the ureter. For example, mice lacking the transcription factor “teashirt 3” fail to develop normal smooth muscle in the ureter and have congenital hydronephrosis without anatomic obstruction.13 Angiotensin plays a key role in initiating the peristalsis, and mice in whom the angiotensin type 1 receptor is knocked out fail to develop a renal pelvis and die of renal failure.9
Angiotensin type 2 receptor gene null mutant mice display congenital anomalies of the kidney and urinary tract. These include unilateral agenesis, unilateral megaureter and hydronephrosis, and pelviureteral junction (PUJ) obstruction and mimic a range of abnormalities found in humans.9 Administration of angiotensin-converting enzyme (ACE) inhibitors throughout pregnancy in humans can cause hypotension and anuria in the baby with histologic features of renal tubular dysplasia. This phenotype is also caused by mutations in genes encoding renin, angiotensinogen, ACE, and angiotensin II receptor type 1. Little is yet known about the genetics of congenital bladder outlet obstruction.13
Other syndromes associated with dysplasia and agenesis in which the mutation is now known include branchio-oto-renal syndrome (EYA1 mutation, transcription factor–like protein), Fraser syndrome (FRAS1 mutation, putative cell adhesion molecule), Kallmann syndrome (X-linked form, KAL1 mutation, cell adhesion molecule; autosomal form, FGFR1 mutation, growth factor receptor), and renal cysts and diabetes syndrome (HNF1 mutation, transcription factor).14
Renal Malformations
Congenitally abnormal kidneys may be large or small, cystic or irregular in outline, and absent or misplaced. These conditions were traditionally discussed on the basis of findings on intravenous urography (IVU), but the findings on computed tomography (CT) and magnetic resonance imaging (MRI) are now increasingly being emphasized.
Large Kidneys
Enlarged kidneys resulting from congenital problems are usually hydronephrotic or cystic. Wilms’ tumor must also be considered. The differential diagnosis in adults of enlarged kidneys, both congenital and acquired, is shown in Chapter 5 (see Fig. 5-1), and the differential diagnosis of cystic kidney disease is discussed further in Chapters 46 and 47.
Irregular Kidneys
Irregularity of the renal outline may result from fetal lobulation or a “dromedary hump,” neither of which has any functional implications. Much more important is the diagnosis of renal dysplasia.
Renal Dysplasia
Table 52-1 presents the range of dysplastic and other malformations of the kidney. Clearly, abnormalities of the ureter, bladder, and urethra are often associated with renal dysplasia.15,16 All types of renal dysplasia can also occur as isolated developmental anomalies. Renal dysplasia, although typically producing small, irregular kidneys, may be cystic or multicystic renal dysplasia.
Table 52-1
Definitions of renal dysplasia and malformation.
| Definitions of Renal Dysplasia and Malformation | |
| Term | Characteristics |
| Renal agenesis | Absence of the kidney or an identifiable metanephric structure |
| Renal aplasia | Severe dysplasia with extremely small kidney, sometimes identifiable only by histologic examination |
| Renal dysplasia | Abnormal differentiation of renal parenchyma with development of abnormal structures, including primitive ducts surrounded by collars of connective tissue, metaplastic cartilage, variety of nonspecific malformations such as preglomeruli of fetal type, and reduced branching of collecting ducts with cystic dilations and primitive tubules |
| Renal hypoplasia | Significantly reduced renal mass and nephron number without evidence of maldevelopment of parenchyma |
| Renal multicystic dysplasia | Severe cystic dysplasia with extremely enlarged kidney full of cystic structures; occurs as an isolated renal lesion in response to ureteral atresia and urethral obstruction; 10% of patients have a family history. |
Renal Hypoplasia (Oligomeganephronia)
Renal hypoplasia is defined as a congenitally small kidney (two standard deviations below the expected mean) that lacks evidence of either parenchymal maldifferentiation (renal dysplasia) or acquired disease sufficient to explain the reduced size. The term is often used loosely, however, and most patients with a small kidney and other malformations will have oligomeganephronia. This is a type of renal hypoplasia resulting from a congenital reduction in the number of nephrons. It results from arrested development of the metanephric blastema at 14 to 20 weeks of gestation with subsequent hypertrophy of glomeruli and tubules in the kidney. The hypertrophy and hyperfiltration result in progressive nephron injury and sclerosis later in life. Oligomeganephronia is recognized on renal biopsy by the large size of the glomeruli and tubules and the small number of glomeruli seen in a good core of renal cortex. It is reported in congenital syndromes caused by mutations in PAX2 and hepatocyte nuclear factor 1β (HNF1β).
Differential Diagnosis of Scarred Kidneys
Dysplasia Versus Reflux
Progressive scarring and renal failure were once considered to be caused by chronic parenchymal infection (so-called chronic pyelonephritis) and were regarded as a consequence of VUR. The 1980s, however, saw a retreat from the paradigm of the primary role of infection, and emphasis was placed on scarring as a result of reflux and the progressive nature of the glomerular lesion associated with glomerular hypertension (or hyperfiltration), so-called reflux nephropathy.3,17,18 The emphasis is changing again to the concept that scarring is often a consequence of renal dysplasia, and that the reflux is a secondary or incidental feature (Fig. 52-2). Thus, irregular kidneys with normal-caliber ureters are more likely to be caused by primary dysplasia, and no evidence of VUR may be seen.
Renal Scarring in Adults
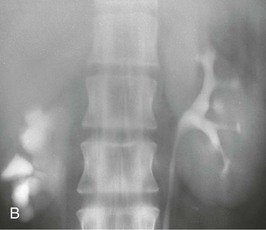
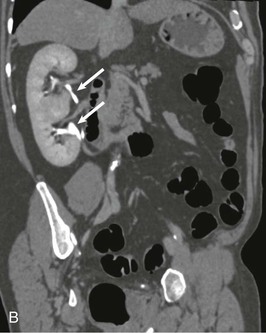
A practical clinical problem is the differential diagnosis of scarred, asymmetric kidneys. With older patients, the differential diagnosis of scarred or “lumpy bumpy” kidneys widens. Often attributed to analgesic nephropathy in the 1970s, this appearance now is often designated “reflux nephropathy.” In older patients, multiple scarring from atheromatous arterial disease and embolization of the kidney is an increasingly important cause of renal failure. The diagnosis has historically been made by the radiologic features on IVU, but in practice, patients often have advanced renal impairment and are unable to excrete enough radiocontrast to delineate the anatomy of the calyces and pelvis and their relationship to the scarring. Newer techniques that include CT and MR urography have supplanted the IVU.19 With urologic conditions, there will be distortion and clubbing of calyces; with other conditions, the calyceal pattern should be normal, except for examples of papillary necrosis (Fig. 52-3). Scarring is best demonstrated by technetium 99m–labeled dimercaptosuccinic acid (99mTc-DMSA) scintigraphy.
Absent Kidneys
Unilateral Renal Agenesis
Complete absence of one kidney occurs in 1 in 520 to 1000 births. It can be familial, and the term hereditary renal aplasia is used by pediatricians. It is an autosomal dominant trait with incomplete penetrance and variable expression and can be associated with bilateral renal agenesis or severe dysplasia. In some families, mutations in uroplakin-IIIa are found.20
Typically, there is no ureter, and the ipsilateral half of the bladder trigone is missing. The remaining kidney is usually hypertrophic, but it may be ectopic, malrotated, or hydronephrotic with a megaureter. The more severe the dysplasia of the remaining kidney, the earlier is the presentation. The ipsilateral testis and seminal tract are usually absent, and in 10% of cases, the adrenal gland is also missing. Girls can have an absent fallopian tube or ovary or malformation of the vagina or uterus. Other associations include imperforate anus and malformations of the vertebrae and cardiovascular system. Agenesis could result from failure in formation of the metanephros or the ureteral bud; however, in association with cloacal abnormalities, the ureteral bud is more likely.
Normality of the single kidney should be confirmed by 99mTc-DMSA scintigraphy, normal isotopic GFR, and absence of proteinuria. If the remaining kidney is not normal, lifelong follow-up is necessary. Ultrasound of the kidneys is recommended in first-degree relatives in all families in whom there is an individual with unilateral or bilateral renal agenesis.
Bilateral Renal Agenesis
Bilateral renal agenesis is lethal. It is associated with pulmonary hypoplasia and a characteristic facial appearance (Potter facies) caused by intrauterine compression, which is a consequence of oligohydramnios. The prevalence is about 1 in 10,000 births, with risk for occurrence in siblings of about 3%, unless there is a family history of agenesis, when risk rises to 15% to 20%.
Misplaced Kidneys
Renal Ectopia, Malrotation, and Crossed Fused Kidneys
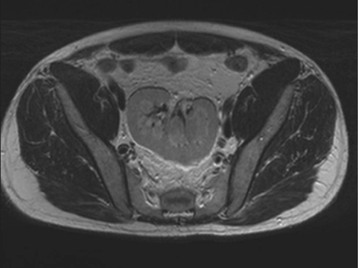
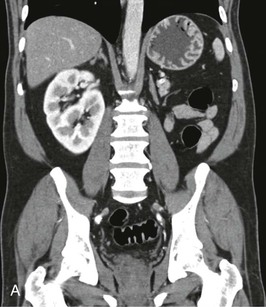
The starting position of the fetal kidney is deep in the pelvis. Kidneys that fail to ascend properly and therefore remain lower than usual occur in 1 in 800 births (Fig. 52-4; see also Fig. 5-16). During development and ascent of the kidney, the renal pelvis comes to face more medially. The most common anomaly is for the pelvis to face forward. The more ectopic the kidney, the more severe is the rotation and more abnormal the appearance. In more than 90% of ectopia, there is fusion of both kidneys. This is now best visualized on CT or MR urography (Fig. 52-5). Symptoms and complications, if any, are caused by associated reflux or PUJ obstruction.
Horseshoe Kidney
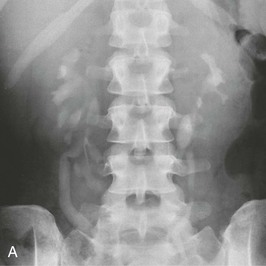
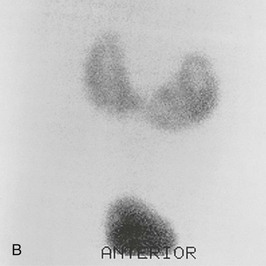
If both kidneys are low, they may join at the lower pole and are usually drained by two ureters (Fig. 52-6). The kidneys lie lower than normal, and further ascent is prevented by the root of the inferior mesenteric artery. Horseshoe kidney occurs in 1 in 400 to 1800 births and is more common in males (2 : 1). Patients present, if at all, with complications of reflux, obstruction, or stone formation.
Calyceal Abnormalities
Hydrocalyx and Hydrocalycosis
Dilated calyces are usually caused by obstruction. Focal dilation can also be caused by congenital infundibular stenosis, extrinsic compression from vessel or tumor, stones, or tuberculosis. If obstruction is excluded, the appearance is likely to be a congenital abnormality and can be an incidental finding. Moreover, if the GFR is normal and the divided function of the kidneys is 50 : 50, surgery to “improve” the anatomy should not be attempted.
Megacalycosis
In megacalycosis, there is bizarre dysplasia of the calyceal system with an increase in the number of calyces. There is no obstruction, and the cause is malformation of renal papillae. Megacalycosis is congenital, usually unilateral, and an incidental finding. It is much more common in males (6 : 1) and occurs only in Caucasians. Bilateral disease is confined to males, and segmental, unilateral disease to females, which suggests an X-linked partially recessive gene with reduced penetrance in females. There may be an associated ipsilateral segmental megaureter, usually affecting the distal third.
Calyceal Diverticulum (Calyceal Cyst)
A calyceal diverticulum is a cavity peripheral to a minor calyx that is not a closed cyst but rather is connected to the calyx by a narrow channel. It is usually an incidental finding in 5 per 1000 IVUs and is best seen on a delayed film (Fig. 52-7). If a calyceal diverticulum is present, symptoms relate to stones or infection within the cavity.
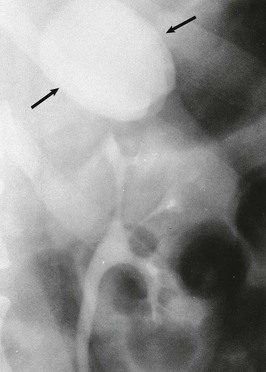
Bardet-Biedl Syndrome
Multiple calyceal clubbing and calyceal diverticula are the characteristic features of the renal dysplasia seen in Bardet-Biedl syndrome (formerly known as Laurence-Moon-Biedl syndrome).21 This autosomal recessive condition is characterized by retinitis pigmentosa, dysmorphic extremities (sometimes with polydactyly), obesity, and hypogonadism. Calyceal malformation is associated with parenchymal dysplasia; renal failure in early adult life is common. Since it was shown that Bardet-Biedl syndrome was caused by a defect of the basal body of ciliated cells,22 mutations in 15 genes coding for different proteins located in the basal body and cilia of the cell have been reported, making the syndrome an archetypal ciliopathy.
Pelviureteral Junction Obstruction
In children, PUJ obstruction is one of the most frequent causes of obstructive uropathy. The condition is usually congenital but can have an acquired mechanical basis caused by stenosis or external compression from adhesions, aberrant lower pole vessels, or kinking of the most proximal ureter. Associated abnormalities are common, and up to 50% of infants have another urologic abnormality, such as contralateral PUJ, contralateral renal dysplastic and multicystic kidney, minor degrees of VUR, and contralateral renal agenesis.
Older children can present with an abdominal mass or with pain in the flank, hematuria secondary to mild trauma, or UTI. Hypertension is unusual but can occur temporarily after surgical correction.
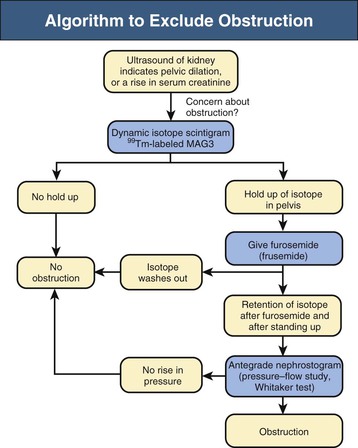
Diagnostic procedures need to differentiate between significant obstruction that requires surgical correction and congenital ectasia of the renal pelvis, in which case surgery is not indicated (Fig. 52-8). Indications for surgical intervention include impairment of renal function, pyelonephritis, renal stones, and pain. Kidneys with good function can generally be left alone, and surgery is indicated only when function is clearly shown to deteriorate.23
Gonadal Dysgenesis
The problems of intersex, gender identity, and micropenis are beyond the scope of this chapter and rarely encountered in adult practice. Patients will be met, however, with ESRD who are phenotypically female but are genotype XY and have mutations of WT1 (Denys-Drash and Frasier syndromes). They have gonadal dysgenesis and must have their streak ovaries removed; otherwise, gonadoblastomas will develop.
Ureteral Abnormalities
The Ureter
The ureter is lined by a multilayered, watertight epithelium (urothelium) surrounded by smooth muscle that generates peristaltic waves distally from pelvis to bladder. The molecular mechanism and growth factors initiating and regulating ureteric budding and branching are increasingly understood, and some genes already implicated in ureteric dysplasia include PAX2, HNF1β, GATA3, ROBO2, and UPK3A.24
Duplex Ureters
Duplication of the ureter and the renal pelvis is a common anomaly, with an incidence of about 1 in 150 births; unilateral duplication is six times more frequent than bilateral. It is more common in girls. If duplication has been detected in a patient, the likelihood of another sibling with duplication rises to 1 in 8.
Pathogenesis
If the ureteral bud bifurcates after its origin from the mesonephric duct but arises at a normal site, an incomplete ureteral duplication with a Y ureter will develop.7 Complete ureteral duplication occurs if there are two ureteral buds, one in the normal location and the other in a low position. The normal bud ends in a correct site on the trigone in the bladder and is nonrefluxing. The lower bud, representing the ureter of the lower pole of the kidney, ends in the bladder as a lateral orifice with a short submucosal tunnel. The lower pole ureter is therefore often associated with VUR, and scarring of the lower pole can result.
If there are two ureteral buds, one with a normal location and one with a high position, the upper ureter is incorporated into the developing bladder, ending more distally and medial to the normal one. Thus, the upper pole ureter ends ectopically, and because of obstruction or dysplasia, there is often severe scarring of the upper pole moiety.
Clinical Manifestations
In most adult patients, ureteral reduplication is asymptomatic and causes no long-term problems. Children with ureteral duplication often have VUR. The spontaneous disappearance of reflux is less common in duplex ureters than in patients with a single ureter.25 Duplex ureters are best diagnosed by CT urogram. PUJ obstruction of the ureter draining the lower pole of the kidney can occur.
Associated conditions, such as ectopic ureters and ureterocele (see later discussion), usually cause problems in early life and therefore have been addressed by adolescence. Upper pole scarring is associated with an ectopic ureter, and lower pole scarring with VUR (Fig. 52-9, A).
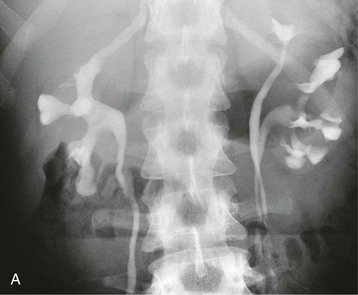
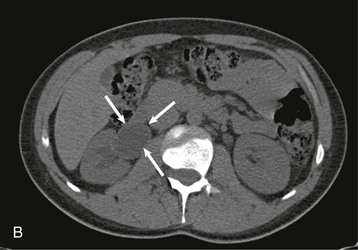
Ectopic Ureters
Ectopic ureters are almost always associated with ureteral reduplication, and 10% are bilateral. There is a female-male ratio of 7 : 1. The ectopic ureter comes from the upper pole and inserts into the bladder more distally and toward the bladder neck, or it opens into the upper urethra. In females, the ureter may end in the urethra, vagina, or vulva, and patients present with incontinence, UTIs, or a persistent vaginal discharge, particularly if the external sphincter is damaged, as during labor.
Ectopic ureters are rare in males and present as UTI. Usually, there is a single ureter associated with a dysplastic kidney, which ends in the posterior urethra, ejaculatory duct, seminal vesicle, or vas. Males are usually continent because the ureter is proximal to the external sphincter.
Ectopic ureters are best visualized by CT or MR urography. A voiding cystourethrogram shows reflux into the lower pole of the kidney in 50% of patients.
Ureterocele
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


