of interstitial fibrosis or scarring. This abnormal matrix is comprised of both a greater abundance of normal interstitial matrix proteins and the de novo appearance of additional matrix proteins. Interstitial fibrosis is accompanied by irreversible tubular damage, ranging from abnormally dilated (ectatic) tubules to atrophic tubules surrounded by abnormally thickened and wrinkled tubular membranes to complete tubular drop-out (often leaving behind the signature “atubular” glomeruli). In parallel, peritubular capillaries are also lost. In some diseases such as chronic allograft rejection, an abnormal process of interstitial lymphangiogenesis has been described, but its specificity and functional significance remain unclear.
TABLE 57.1 Chronic Tubulointerstitial Nephritis Classification | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
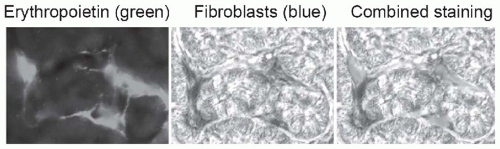 FIGURE 57.1 Peritubular fibroblasts produce erythropoietin. Using a mouse line that was genetically engineered to express green fluorescent protein-labeled erythropoietin, and kidney Cre-labeled fibroblasts that are detectable by beta galactosidase staining (blue), peritubular fibroblasts are identified as the source of erythropoietin. Loss of this function explains why anemia may be more severe in patients with chronic kidney disease due to chronic tubulointerstitial nephritis. (From Asada N, Takase M, Nakamura J, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 2011;121:3981, with permission.) (See Color Plate.) |
change in interstitial cellularity is the appearance of an infiltrate of mononuclear cells. These cells are primarily of bone marrow origin.
TABLE 57.2 Histopathologic Features of Acute and Chronic Tubulointerstitial Nephritis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Fanconi syndrome. Low molecular weight proteins such as β2-microglobulin may not be properly reabsorbed, leading to tubular proteinuria. Distal tubular dysfunction manifests as renal sodium wasting, hyperkalemia, and nonanion gap metabolic acidosis. Collecting duct dysfunction leads to renal concentrating defects including features of diabetes insipidus and countercurrent exchange washout resulting in polyuria. Most TIN affects multiple sites of the nephron simultaneously, but to varying degrees. Hypertension, severe proteinuria, and edema are not usually characteristic of TIN in the early stage, but may develop later as progressing chronic kidney disease (CKD) with glomerular sclerosis. In addition to tubular dysfunction, anemia may be found disproportionally compared to the change in GFR if erythropoietin-producing peritubular cells (Fig. 57.1) are damaged early in the disease. Bone disease may also be prominent, as a result of chronic phosphate wasting caused by proximal tubular dysfunction.
 FIGURE 57.2 Interstitial fibrosis: detection and correlation with renal functional loss. The fibrotic or scarred interstitium contains several extracellular matrix proteins, the most abundant being fibrillar collagens such as collagen III (A,B). Routine renal biopsy staining with Masson trichrome reacts with collagen to produce a green-blue color (C). Quantitative pathologic research studies often use picrosirius red staining, which is specific for cross-linked collagen fibrils (polarized image shown in D). The key structural change that underlies the loss of renal function in all chronic kidney diseases is tubular atrophy (upper graph) which is closely associated with interstitial fibrosis severity (lower graph). (A and B are from Jones CL, Buch S, Post M, et al. Pathogenesis of interstitial fibrosis in chronic purine aminonucleoside nephrosis.Kidney Int. 1991;40:1020. Upper graph is from Mackensen-Haen S, Bohle A, Christensen J, et al. The consequences for renal function of the interstitium and changes in the tubular epithelium of the cortex and medulla in various diseases. Clin Nephrol. 1992;37;70. Lower graph is from Schainuck LI, Striker GE, Cutler RE, et al. Structural-functional correlations in renal diseases: Part II: the correlations.Human Pathol. 1970;1:631. All are reproduced with permission.) (See Color Plate.) |
component C3, and osteopontin (OPN) activate capillary endothelial cells, leading to increased capillary permeability, recruitment of leukocytes into the interstitium, and activation of myofibroblast precursors. Inflammatory macrophages secrete diverse proinflammatory and profibrotic products that perpetuate injury and promote scarring. This process unleashes a cascade of inflammatory and fibrogenic signals within the interstitium. Some key molecules including fibrinogen, complement components C3a and C5a, tissue plasminogen activator (tPA), and oxidized albumin may arrive by leakage from the plasma, whereas others, such as the major fibrogenic factor transforming growth factor β (TGF-β), connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), tumor necrosis factor α (TNF-α), endothelin-1, angiotensin-II, placental growth factor, and angiopoietin-2, appear to be produced locally. Together, these factors activate fibroblasts, and promote their transformation into α-SMA positive myofibroblasts. The activated myofibroblasts produce collagen I, collagen III, fibronectin, and other matrix proteins, which accumulate in the interstitial space. The fibrotic process culminates in death of tubular cells and peritubular capillaries, leading to ablation of the entire nephron (Fig. 57.3).9,10,11,12
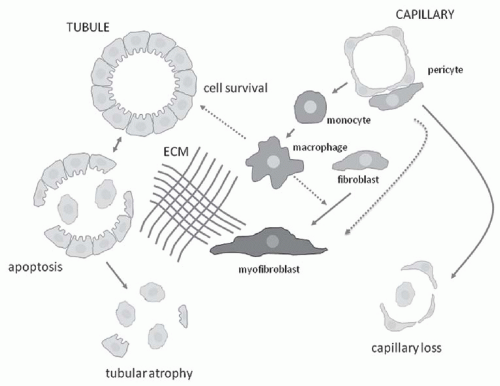 FIGURE 57.3 Schematic summary of the key cellular events contributing to the pathogenesis of chronic tubulointerstitial nephritis. Inflammatory macrophages are primarily recruited from the circulating pool of peripheral blood monocytes, under the direction of chemotactic signals derived from endothelial cells and damaged tubules and facilitated by increased capillary permeability. Functionally distinct macrophage subpopulations either propagate injury or promote tissue repair (including fibrosis) by releasing a variety of cytokines, growth factors, and other soluble products. A population of myofibroblasts appears de novo in the interstitium where they synthesize the majority of the extracellular matrix (ECM) proteins responsible for interstitial scarring. The primary origin of the myofibroblasts is still controversial; resident interstitial fibroblasts and capillary pericytes are considered most likely during active fibrogenesis. Through a variety of mechanisms, including hypoxia and oxidant stress, interstitial capillaries disappear and tubular epithelia undergo apoptotic death in parallel with progressive fibrosis. |
renal injury may expose neoantigens that trigger a secondary antigen-driven immune response to propagate injury, but this hypothesis has been difficult to test using experimental models. Candidate neoantigens have not been identified, and thus this paradigm remains hypothetical.
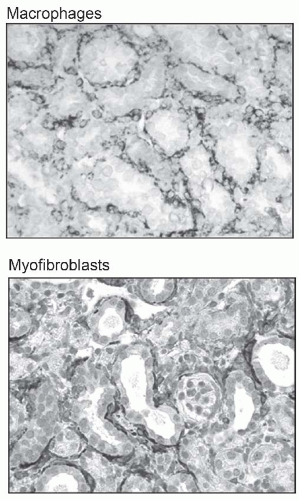 FIGURE 57.4 Interstitial cell mediators of chronic tubulointerstitial nephritis (TIN). Interstitial hypercellularity is a feature of chronic TIN, characterized by the presence of two distinct cell populations: lymphohematopoietic cells, with macrophages in particular (shown by CD68 staining in the upper photomicrograph) known to play an important pathogenic role, and myofibroblasts (shown by alpha smooth muscle actin staining in the lower photomicrograph). Limited human biopsy data show a significant relationship between interstitial myofibroblast density and renal prognosis. (The upper image is from Yamaguchi I, Tchao BN, Burger NL, et al. Vascular endothelial cadherin modulates renal interstitial fibrosis. Nephron Exp Nephrol. 2011; 120:e20, with permission.) |
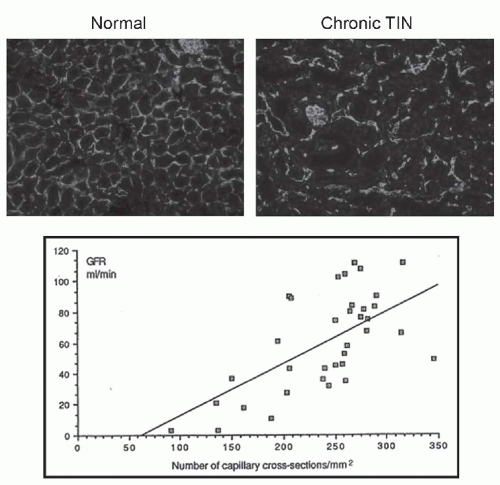 FIGURE 57.5 Interstitial capillary rarefaction is a feature of chronic tubulointerstitial nephritis (TIN). Using CD31 as an endothelial cell marker, the decrease in interstitial capillary cell density in chronic TIN is illustrated by the photomicrographs. In a study of human kidney biopsies, the extent of capillary loss was shown to correlate with the decline in glomerular filtration rate. (The photomicrographs are from Yamaguchi I, Tchao BN, Burger NL, et al. Vascular endothelial cadherin modulates renal interstitial fibrosis.Nephron Exp Nephrol. 2011;120:e20 and the graph is from Serón D, Alexopoulos E, Raftery MJ, et al. Number of interstitial capillary cross-sections assessed by monoclonal antibodies: relation to interstitial damage. Nephrol Dial Transplant. 1990;5:889, both with copyright permission.) |
urokinase-type plasminogen activator (uPA), tPA, and plasmin have been investigated as alternative candidates, but tPA and plasmin were found to promote fibrosis and uPA had no effect, despite the fact that the inhibitor PAI-1 is a potent fibrosis-promoting molecule. The latter effect may best be explained by PAI’s ability to enhance macrophage and myofibroblast recruitment in the interstitium. The urokinase receptor (uPAR) attenuates myofibroblast recruitment and fibrosis, and acts in conjunction with its coreceptor LDL receptor-related protein (LRP) to regulate fibroblast proliferation and extracellular signal-regulated kinase (ERK) signaling.44 Recent studies have focused on the role of an uPAR coreceptor, uPAR-associated protein (uPARAP), also known as the mannose receptor 2 (Mrc2) and Endo180. This receptor is expressed by interstitial macrophages and myofibroblasts and serves as a collagen endocytic receptor that delivers interstitial collagens to lysosomes for degradation by cathepsins.43 Renal fibrosis is significantly worse in mice with genetic Mrc2 deficiency.
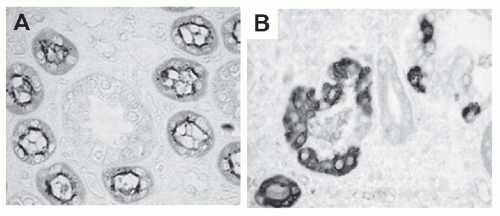 FIGURE 57.6 Familial chronic tubulointerstitial nephritis associated with mutations in the uromodulin (UMOD) gene, which encodes a protein normally expressed on the apical membrane of the thick ascending limb of the loop of Henle (shown on left). These mutations result in an abnormal UMOD protein that is trapped within the endoplasmic reticulum (right photograph). (From Dahan K, Devuyst O, Smaers M, et al. A cluster of mutations in the UMOD gene causes familial juvenile hyperuricemic nephropathy with abnormal expression of uromodulin. J Am Soc Nephrol. 2003;14:2883, with permission.) |
cells, causing additional damage to the RAS and leading to nephron dropout and progressive renal failure. Patients typically present with early-onset anemia due to low erythropoietin production, mild hyperkalemia, and low-normal blood pressure. A history of gout in some affected family members should raise the suspicion of this disorder. Plasma renin, aldosterone, and erythropoietin levels are low; fractional urate excretion is reduced; and kidney biopsies show chronic TIN, although none of these findings alone confirms the diagnosis. Mutational analysis of the REN gene is required. Treatment is supportive. CKD typically develops in the third or fourth decade and progresses slowly.
diagnosis is typically confirmed by the presence of elevated cystine levels in peripheral blood leukocytes, measured in a reference laboratory. Since the cystinosis gene (CTNS) was cloned in 1998, over 90 mutations have been reported in the United States and northern Europe—approximately 40% have a homozygous 57 kb deletion.
 FIGURE 57.7 Interstitial inflammation is a pathogenic feature of polycystic kidney disease. In a mouse model of autosomal dominant polycystic kidney disease, macrophages (green) are seen lining cystic spaces (upper left). When macrophages were experimentally depleted (upper right), renal parenchyma was better preserved (lower left), and kidney function estimated by blood urea nitrogen levels was significantly better in the macrophage depleted (—) mice, shown in the lower right graph. (From Karihaloo A, Koraishy F, Huen SC, et al. Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2011;22:1809, with permission.) (See Color Plate.) |
with photophobia that requires treatment with topical cysteamine. Neuromuscular involvement and pancreatic insufficiency often develop in the older patients.
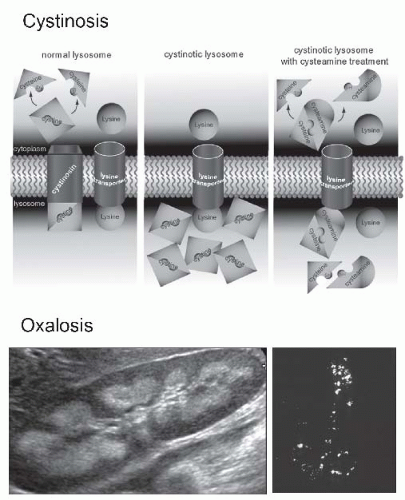 FIGURE 57.8 Inherited metabolic diseases cause chronic tubulointerstitial nephritis. The upper drawing illustrates the abnormal function of the lysosomal membrane in patients with autosomal recessive cystinosis due to a mutation in the CTNS gene that encodes the cystine transporter cystinosin. In the absence of cystinosin, cystine accumulates in lysosomes and contributes to some of the associated tissue pathologies. An elevated peripheral blood leukocyte cystine level is diagnostic. The drug cysteamine provides an alternative pathway for cystine exit from lysosomes by forming a cysteine-cysteamine dimer that is transported by an alternative (system c) lysosomal transporter. Though not yet definitively identified, it has been suggested that system c is the lysine transporter. (From Wilmer MJ, Schoeber JP, van den Heuvel P, et al. Cystinosis: practical tools for diagnosis and treatment.Pediatr Nephrol. 2011;26:205, with permission.) Primary infantile oxalosis is associated with aggressive chronic tubulointerstitial nephritis due to the deposition of calcium oxalate in renal tubules and the interstitium, which may be detected as nephrocalcinosis on the renal ultrasound (lower left) or the actual deposits can be visualized by polarized light microscopic examination of kidney tissue (lower right). (See Color Plate.) |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


