Cardiac Failure and the Kidney
William T. Abraham
Robert W. Schrier
The kidney plays a central role in the sodium and water retention and edema formation associated with cardiac failure. Heart failure, like liver disease and the nephrotic syndrome, represents another edematous state in which renal sodium and water retention is observed despite an excess of total body sodium and water. This finding of continued renal sodium and water retention despite total body sodium and water excess, in part, defines the clinical syndrome of heart failure. In this regard, the pathophysiology of heart failure has been described as a cardiorenal syndrome, where left ventricular systolic and/or diastolic dysfunction leads to renal sodium and water retention that in turn produces the clinical syndrome of heart failure. Manifestations of cardiac failure are almost always associated with fluid volume retention resulting in hemodynamic and clinical congestion, where the former is measured as elevated ventricular filling pressures and the latter is seen as congestive signs and symptoms. Although an abnormal cardiac output initiates renal sodium and water retention, as will be discussed later in this chapter, most of the cardinal signs and symptoms of heart failure are attributable to fluid retention rather than to an abnormal cardiac output (Table 67.1).
Moreover, worsening fluid retention is the proximate cause of heart failure hospitalization (i.e., morbidity) in nearly 90% of cases.1,2 As will be discussed in subsequent text of this chapter, renal dysfunction as measured simply by elevated blood urea nitrogen (BUN) and/or serum creatinine portends a very poor prognosis in both acutely decompensated patients and patients with chronic heart failure.3,4,5,6 Consequently, the kidney provides a sensitive bioassay for prognosis in patients with heart failure. This observation underscores the importance of cardiorenal interactions in the natural history of heart failure. This chapter reviews the mechanisms of edema formation and sodium and water retention associated with cardiac failure, discusses the clinical implications of cardiorenal interactions in heart failure, and reviews current and future treatment options.
THE MECHANISM OF EDEMA FORMATION
Edema is a clinical sign that indicates an increase in the volume of sodium and water in the interstitial space. This increase in interstitial-space volume is caused by an alteration of the Starling forces that govern the transfer of fluid from the vascular compartment into the surrounding tissue spaces.7 Edema may result from local factors such as an obstruction of lymphatic or venous flow. However, the type of edema considered in this chapter reflects a generalized disturbance of sodium and water balance and is associated with a net increase in extracellular fluid (ECF) volume, a situation that is usually not present when edema results from a local disruption of normal capillary mechanisms. Generalized edema results when altered Starling forces affect all capillary beds. The development of generalized edema thus indicates a widespread disturbance in the normal balance between tissue capillary and interstitial hydrostatic and colloid osmotic pressures, which control the distribution of ECF between the vascular and extravascular (interstitial) compartments. In edematous disorders such as cardiac failure, sodium and water retention by the kidney leads to the progressive expansion of the ECF volume and alteration of the Starling forces that subsequently result in edema formation.
Transcapillary solute and fluid transport consists of two types of flow: convective and diffusive. Bulk water movement occurs via convective transport induced by the imbalance between transcapillary hydraulic pressure and colloid osmotic pressure.7 Transcapillary hydraulic pressure is influenced by a number of factors, including systemic arterial and venous blood pressures, regional blood flow, and the resistances imposed by the precapillary and postcapillary sphincters. Cardiac output, intravascular volume, and systemic vascular resistance, in turn, determine systemic arterial blood pressure. Systemic venous pressure is determined by right atrial pressure, intravascular volume, and venous capacitance. These latter hemodynamic parameters are largely determined by sodium and water balance and by various neurohormonal factors. For
example, right atrial pressure or right ventricular preload is modulated both by changes in the intravascular volume, which are largely determined by the kidney, and alterations in venous capacitance, which are governed in part by neuroendocrine mechanisms such as the sympathetic nervous system, the renin-angiotensin system, the nonosmotic release of arginine vasopressin (AVP), and the natriuretic peptides. As discussed in this chapter, activation of these two mechanisms (i.e., renal sodium and water retention and neurohormonal activation), which may influence transcapillary hydraulic and oncotic pressures, is observed with cardiac failure.
example, right atrial pressure or right ventricular preload is modulated both by changes in the intravascular volume, which are largely determined by the kidney, and alterations in venous capacitance, which are governed in part by neuroendocrine mechanisms such as the sympathetic nervous system, the renin-angiotensin system, the nonosmotic release of arginine vasopressin (AVP), and the natriuretic peptides. As discussed in this chapter, activation of these two mechanisms (i.e., renal sodium and water retention and neurohormonal activation), which may influence transcapillary hydraulic and oncotic pressures, is observed with cardiac failure.
TABLE 67.1 Common Signs and Symptoms of Congestive Heart Failure | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Several mechanisms are capable of minimizing edema formation or diminishing the transudation of solute and water across the capillary bed. In several vascular beds, the local transcapillary hydraulic pressure gradient exceeds the opposing colloid osmotic pressure gradient throughout the length of the capillary bed, so that filtration occurs across its entire length.8 Filtered fluid consequently must return to the circulation via lymphatics. Increased lymphatic drainage and the ability of lymphatic flow to increase may thus be seen as one protective mechanism that minimizes edema formation. Other protective mechanisms that reduce interstitial fluid accumulation include precapillary vasoconstriction, increased net filtration with a resultant rise in intracapillary plasma protein concentration, and increased interstitial fluid volume with a resultant augmentation of tissue hydraulic pressure. For example, increased net filtration itself, such as that associated with hypoalbuminemia and the resultant decreased plasma oncotic pressure, leads to a dissipation of capillary hydraulic pressure, a dilution of interstitial fluid protein concentration, and a corresponding rise in intracapillary protein concentration, all of which alter the balance of the Starling forces to mitigate further interstitial fluid accumulation.
These buffering factors directed against interstitial fluid accumulation may explain why, in patients with congenital analbuminemia, positive sodium and water balance, and edema formation do not occur consistently and sodium loads are excreted.9 Because the continued loss of intravascular fluid volume to the interstitial space without renal sodium and water retention may result in the cessation of interstitial fluid formation, the presence of generalized edema, therefore, implies concomitant renal sodium and water retention. This is unquestionably the case in cardiac failure, as well as in liver disease and the nephrotic syndrome. The disturbances in microcirculatory hemodynamics associated with edema and expansion of the ECF volume are described in Table 67.2.
THE MECHANISMS OF FLUID RETENTION IN CARDIAC FAILURE
Cardiac failure may be defined as the inability of the heart to deliver enough blood to peripheral tissues to meet metabolic demands. In the case of low-output cardiac failure, a decrease in cardiac output initiates a complex set of compensatory mechanisms in an attempt to maintain circulatory integrity. The adjustments that serve to stabilize cardiac performance and arterial perfusion in such patients include increases in plasma volume, atrial and ventricular filling pressures, peripheral vasoconstriction, and cardiac contractility
and heart rate. The retention of sodium and water is a major renal compensation for a failing myocardium, but it also accounts to a great extent for the familiar clinical syndrome of heart failure, which consists of pulmonary and/or peripheral edema and exercise intolerance. In fact, the inability to excrete a sodium load has been used as an index of the presence of heart failure,10 and a defect in water excretion is regularly encountered in such patients.11
and heart rate. The retention of sodium and water is a major renal compensation for a failing myocardium, but it also accounts to a great extent for the familiar clinical syndrome of heart failure, which consists of pulmonary and/or peripheral edema and exercise intolerance. In fact, the inability to excrete a sodium load has been used as an index of the presence of heart failure,10 and a defect in water excretion is regularly encountered in such patients.11
TABLE 67.2 Disturbances in Microcirculatory Hemodynamics Associated with Edema and Expansion of Extracellular Fluid Volume | ||||
|---|---|---|---|---|
|
Classically, two theories have attempted to explain how the kidney becomes involved in renal sodium and water retention of heart failure. According to the “backward failure” hypothesis advanced by Hope12 and Starling,7 central venous pressure rises and then peripheral venous pressure rises as the cardiac pump fails. With this increase in peripheral venous pressure, the hydraulic pressure in the capillaries exceeds opposing forces and causes the transudation of fluid from the intravascular compartment to the interstitial space, and thus the development of edema. This loss of intravascular fluid volume then signals the kidney to retain sodium and water in an attempt to restore the circulating volume to normal. The “forward failure” theory states that as the heart fails, there is inadequate perfusion of the kidney, resulting in decreased sodium and water excretion.13 As will become apparent from the following discussion, both an increase in central venous pressure, or “backward failure,” and a decrease in cardiac output, or “forward failure,” may contribute to the sodium and water retention of low-output cardiac failure via systemic and renal hemodynamic effects and through the activation of various vasoconstrictor and antinatriuretic neuroendocrine systems. According to our unifying hypothesis of body fluid-volume regulation,14,15,16,17,18,19,20,21 neurohormonal activation plays a central role in the efferent limb of the sodium and water retention in cardiac failure, liver disease, and the nephrotic syndrome, whereas the afferent limb of this volume regulatory system is initiated by altered systemic hemodynamics. The following discussion addresses this unifying hypothesis of body fluid-volume regulation and the afferent and efferent mechanisms for sodium and water retention in edematous disorders, in particular, heart failure.
Afferent Mechanisms for Renal Sodium and Water Retention in Heart Failure
The kidney alters the amount of dietary sodium excreted in response to signals from volume receptors and chemoreceptors in the circulation. These receptors may affect kidney function by altering renal sympathetic nerve activity and changing levels of circulating hormones with vasoactive and nonvasoactive (e.g., direct sodium-retaining) effects on the kidney. Important “effector” hormones include angiotensin II (AT-II), aldosterone, AVP, endothelin, nitric oxide (NO), prostaglandins (PGs), and the natriuretic peptides, especially atrial and brain natriuretic peptides (ANP and BNP, respectively). Both high- and low-pressure baroreceptors as well as cardiac and hepatic chemoreceptors have been implicated in the activation of these neurohormonal systems.
High-Pressure Baroreceptors
In humans, evidence for the presence of volume-sensitive receptors in the arterial circulation originated from observations in patients with traumatic arterial-venous (AV) fistulae.22 Closure of AV fistulae is associated with a decreased rate of emptying of the arterial blood into the venous circulation, as demonstrated by closure-induced increases in diastolic arterial pressure and decreases in cardiac output. This results in an immediate increase in renal sodium and water excretion without changes in either glomerular filtration rate (GFR) or renal blood flow.22 This observation implicates the “fullness” of the arterial vascular tree as a “sensor” in modulating renal sodium and water excretion. In fact, the fullness of the arterial vascular compartment, or the so-called effective arterial blood volume (EABV),23 has been proposed as a major determinant of renal sodium and water handling according to the unifying hypothesis of body fluid volume regulation.14,15,16,17,18,19,20,21
The EABV is a measure of the adequacy of arterial blood volume to “fill” the capacity of the arterial circulation. Normal EABV exists when the ratio of cardiac output to peripheral vascular resistance maintains venous return and cardiac output at normal levels. Arterial or high-pressure volume receptors, therefore, may be stimulated when either cardiac output falls or peripheral vascular resistance diminishes to such an extent that the arterial circulation is no longer effectively “full” (Fig. 67.1). Therefore, in the case of low-output cardiac failure, it is the diminution of cardiac output that is perceived by the arterial circulation as inadequate to maintain EABV. In high-output cardiac failure, decreased peripheral vascular resistance may serve as the signal for arterial underfilling.14,15,16,17,18,19,20,21 The concept of arterial underfilling in low- and high-output cardiac failure is discussed in the following paragraphs.
Studies using one model of low-output cardiac failure— constriction of the vena cava in the dog—support the notion that a fall in cardiac output may be a primary stimulus for sodium and water retention by the kidney. Using this model, Schrier and associates24,25,26 showed that constriction
of the thoracic inferior vena cava (TIVC) is associated with a decrease in cardiac output, arterial pressure, and urinary sodium excretion, even when renal perfusion pressure and renal venous pressure were held constant. Of note, renal denervation and adrenalectomy did not abolish this antinatriuresis. Furthermore, sodium retention did not correlate with changes in GFR or renal vascular resistance. Constriction of the superior vena cava to cause a decrease in cardiac output similar to that observed in the TIVC studies resulted in a similar decrease in urinary sodium excretion despite the absence of concomitant hepatic, renal, and abdominal venous congestion. These findings support the hypothesis that the kidney decreases sodium excretion in response to a decrease in cardiac output and the associated arterial underfilling.
of the thoracic inferior vena cava (TIVC) is associated with a decrease in cardiac output, arterial pressure, and urinary sodium excretion, even when renal perfusion pressure and renal venous pressure were held constant. Of note, renal denervation and adrenalectomy did not abolish this antinatriuresis. Furthermore, sodium retention did not correlate with changes in GFR or renal vascular resistance. Constriction of the superior vena cava to cause a decrease in cardiac output similar to that observed in the TIVC studies resulted in a similar decrease in urinary sodium excretion despite the absence of concomitant hepatic, renal, and abdominal venous congestion. These findings support the hypothesis that the kidney decreases sodium excretion in response to a decrease in cardiac output and the associated arterial underfilling.
 FIGURE 67.1 Peripheral vascular resistance and cardiac output as the determinants of arterial filling or the “effective arterial blood volume.” Here, either a decrease in vascular resistance or diminished cardiac output results in decreased fullness of the arterial circulation with unloading of high-pressure volume receptors and activation of various neurohormonal responses (see text). |
Migdal et al.27 questioned this hypothesis by comparing the renal response in three different models of experimental heart failure. Specifically, they compared models of TIVC constriction, pulmonary artery occlusion (which is similar to caval constriction except that right-sided heart pressures are increased rather than decreased), and acute left ventricular infarction, another model of low-output heart failure but with increased left-sided heart pressures. This investigation demonstrated that with comparable decrements in cardiac output in all three models, only the TIVC constriction animals exhibited antinatriuresis. The authors concluded that low cardiac output per se is not the afferent signal for sodium retention in low-output heart failure. These authors27 and others28 suggested that in some way, decreased right-sided heart pressure mediates the antinatriuresis.
An alternative interpretation of the findings of Migdal et al.27 is that a decrease in cardiac output is a stimulus for renal sodium and water retention, but an acute rise in atrial or ventricular end-diastolic pressures, in animals with acute pulmonary hypertension or acute left ventricular infarction, with the release of the natriuretic peptides ANP and BNP, initially obscures this effect. Support for this interpretation may be found in a report from Lee et al.,29 who examined sodium excretion in two models of low-output heart failure in the dog, acute heart failure produced by rapid ventricular pacing, and a TIVC constriction model. Similar to the animals in the study of Migdal et al., the dogs with TIVC constriction demonstrated diminished cardiac outputs and arterial pressures without an increase in atrial pressures or plasma ANP level but with avid renal sodium retention. Of note, plasma renin activity (PRA) and plasma aldosterone concentrations were substantially elevated in these TIVC-constriction animals. In the case of pacing-induced heart failure, cardiac output and arterial pressure were similarly decreased, whereas atrial pressures and the plasma ANP concentration were significantly increased. In the animals with elevated rather than normal circulating ANP concentrations, urinary sodium excretion was maintained and PRA and plasma aldosterone concentrations were not increased. Finally, dogs with TIVC constriction were given exogenous ANP to achieve circulating concentrations comparable to that seen in the pacing-induced heart failure animals. Exogenous administration of ANP to such levels prevented sodium retention, renal vasoconstriction, and activation of the renin-angiotensin-aldosterone system. These observations support the notion that decreased cardiac output is a stimulus for renal sodium retention in heart failure and suggest an important role for the natriuretic peptides in acutely attenuating this renal response. A further discussion of the role of ANP and BNP in heart failure is presented elsewhere in this chapter.
Other experimental evidence supports a role for diminished cardiac output as a determinant of sodium and water retention in heart failure. Rats with small-to-moderate myocardial infarctions and decreased cardiac outputs exhibit decreased fractional sodium excretion despite normal right and left ventricular end-diastolic pressures.30 Using the model of TIVC constriction, Priebe et al.31 demonstrated that the renal retention of sodium and water was reduced markedly when cardiac output was restored to normal by autologous blood transfusions. Moreover, a reduction of pressure or stretch at the carotid sinus, like that produced by decreased cardiac output or arterial hypotension, activates the sympathetic nervous system and promotes renal sodium and water retention.32,33 Pharmacologic or surgical interruption of sympathetic afferent neural pathways emanating from high-pressure baroreceptor sites also inhibits the natriuretic response to volume expansion.25,26,34,35,36,37,38 High-pressure baroreceptors also appear to be important factors in regulating the nonosmotic release of AVP, thereby affecting renal water excretion.39,40 Finally, the juxtaglomerular apparatus, an arterial baroreceptor located in the afferent arterioles within the kidney, has been implicated in the modulation of renal renin release32,41,42 and thus may stimulate increases in circulating AT-II and aldosterone, both of which promote sodium retention by the kidney.
Low cardiac output cannot be the only cause of sodium and water retention in heart failure, because diminished renal sodium and water excretion is also observed in states of high-output cardiac failure. In heart failure secondary to beriberi, anemia, thyrotoxicosis, or AV fistulae, cardiac output is increased as a consequence of a decrease in peripheral vascular resistance. This decrease in vascular resistance diminishes EABV (i.e., causes arterial underfilling) and serves as the stimulus for neurohormonal activation and renal sodium and water retention in these instances of high-output heart failure.14,15,16,17,18,19,20,21 As noted already in humans22 and dogs,43 closure of an AV fistula causes increased sodium excretion, whereas opening an AV fistula decreases urinary sodium excretion. These changes in renal sodium excretion correlate with changes in arterial pressure and peripheral vascular resistance rather than GFR or renal blood flow, supporting the importance of arterial circulatory “fullness” as a determinant of the renal response to heart failure.
These observations of decreased sodium and water excretion in both low- and high-output cardiac failure support the theory that arterial underfilling initiates reflex stimuli for the kidneys to retain sodium and water. In this regard, high-pressure baroreceptors in the carotid sinus, aortic arch, left
ventricle, or the juxtaglomerular apparatus may comprise an important part of this reflex loop. Although these data support a role for arterial underfilling as the primary stimulus of the renal sodium and water retention of heart failure, low-pressure baroreceptors also may play an important role.
ventricle, or the juxtaglomerular apparatus may comprise an important part of this reflex loop. Although these data support a role for arterial underfilling as the primary stimulus of the renal sodium and water retention of heart failure, low-pressure baroreceptors also may play an important role.
Low-Pressure Baroreceptors
In addition to the high-pressure arterial baroreceptors, the venous side of circulation seems to be a logical place for receptors sensitive to changes in blood volume to be found. In fact, 85% of blood volume may be found in the venous circulation, whereas just 15% of circulatory volume resides in the arterial circulation.44 Although the smaller arterial blood volume may result in a higher sensitivity to detect blood volume changes, the larger amount of venous blood volume also may constitute an important component of the body fluid-volume regulatory system.
The atria of the heart are highly distensible and densely populated with nerve endings that are sensitive to small changes in passive distention.45 Similar afferent low-pressure volume receptors may also be found in the pulmonary vasculature.46 Increased filling of the thoracic vascular and cardiac atria would be expected to signal the kidney to increase urinary sodium excretion in order to return the blood volume to normal. As expected, maneuvers that increase this thoracic or “central” blood volume, such as weightlessness, negative-pressure breathing, head-out water immersion, recumbency, and exposure to cold, all produce a natriuresis.47,48,49,50,51,52 Similarly, measures that decrease intrathoracic blood volume, including positive-pressure breathing, upright posture, and the application of tourniquets to the lower extremities, result in renal sodium retention.49,53,54 Therefore, effective “central” blood volume, in addition to EABV, may serve as the afferent stimulus for the regulation of renal sodium and water excretion.
Considerable evidence implicates the left atrium as an important site of low-pressure receptors.55,56,57 It is believed that changes in pressure or distention within the left atrium modulate electrical activity of the atrial receptors, which in turn may regulate renal sympathetic nerve activity. Left atrial nerves, therefore, can alter blood volume through changes in sodium excretion57,58,59 as well as solute-free water excretion by influencing AVP release.60,61,62 Acutely increasing left atrial volume by inflation of a balloon within the left atrium results in increased urinary volume excretion,56 whereas hypotensive hemorrhage63,64 and atrial tamponade65 cause decreased atrial volume and diminish urine volume. However, in the setting of chronic heart failure, renal sodium and water retention occur despite left atrial distention and, frequently, loading of the other central baroreceptors (pulmonary veins, right atrium). Therefore, in chronic heart failure, diminished cardiac output with arterial underfilling may exert the predominant effect via the unloading of high-pressure arterial baroreceptors. Chronic studies in animals employing either experimental tricuspid insufficiency66 or right atrial distention with an inflatable balloon67 support this hypothesis. In these animal models, the increase in right atrial pressure was associated with avid renal sodium retention rather than the expected natriuresis. However, a concomitant fall in cardiac output could explain the sodium retention. Alternatively, alterations in cardiopulmonary baroreceptor function may occur in chronic but not acute heart failure.
Zucker et al.68 demonstrated that the inhibition of renal sympathetic nerve activity seen during acute left atrial distention is lost during chronic heart failure in the dog. Moreover, a decrease in cardiac preload fails to produce the expected parasympathetic withdrawal and sympathetic activation in humans with heart failure.69,70,71 Nishian et al.71 described paradoxical forearm vasodilation and hemodynamic improvement during acute unloading of cardiopulmonary baroreceptors in patients with severe chronic heart failure. This paradoxical response to lower body negative pressure was associated with static plasma norepinephrine levels,71 rather than the expected increase in plasma norepinephrine concentrations, further demonstrating this altered response to low-pressure baroreceptor unloading in heart failure. These observations confirm those made in heart failure patients during other forms of orthostatic stress.69,70 These findings are also consistent with the observation of a strong positive correlation between left atrial pressure and coronary sinus norepinephrine, a marker of cardiac adrenergic activity, in patients with chronic heart failure.72 Finally, Fonarow et al.73 have shown that a reduction in left ventricular filling pressure rather than an increase in cardiac output during tailored hemodynamic management of heart failure improves survival over a 2-year period of follow-up. Taken together, these findings suggest that the normal inhibitory control of sympathetic activation accompanying increased atrial pressures is lost in heart failure patients and somehow may be converted to a stimulatory signal.
Cardiac and Pulmonary Chemoreceptors
In the heart and lungs, both vagal and sympathetic afferent nerve endings respond to a variety of exogenous and endogenous chemical substances, including capsaicin, phenyldiguanidine, bradykinin, substance P, and PGs. Baker et al.74 demonstrated stimulation of sympathetic afferent nerve endings by bradykinin in the heart of the cat. In conscious dogs, the administration of PGE2 and arachidonate inhibited the cardiac baroreflex.75 Moreover, Zucker et al.76 showed that PGI2 attenuates the baroreflex control of renal nerve activity via an afferent vagal mechanism. Because substances such as bradykinin and PGs may circulate at increased concentrations in subjects with heart failure,77 it is possible that altered central nervous system input from chemically sensitive cardiac or pulmonary afferents contributes to the neurohormonal activation and sodium retention of chronic heart failure. This possibility may have important implications for the treatment of heart failure, because commonly prescribed medications such as angiotensin-converting enzyme (ACE) inhibitors may alter circulating bradykinin and PG levels. At the present
time, however, the exact roles of these hormones and cardiac and pulmonary chemoreceptors in heart failure are incompletely understood.
time, however, the exact roles of these hormones and cardiac and pulmonary chemoreceptors in heart failure are incompletely understood.
Hepatic Receptors
Theoretically, the liver should be in an ideal position to monitor dietary sodium intake and thus adjust urinary sodium excretion. Indeed, when compared with peripheral venous administration, infusion of saline solution into the portal circulation was reported to result in greater natriuresis.78,79 Similarly, the increment in urinary sodium excretion has been claimed to be greater when the sodium load is given orally than when given intravenously.80,81,82 In addition, the pathophysiologic retention of sodium in patients with severe liver disease is also consistent with an important role for the liver in the control of sodium excretion. However, some investigators83,84 were unable to demonstrate a difference in sodium excretion between animals infused with 5% sodium chloride systemically and animals receiving the same solution via the portal vein. Moreover, Obika et al.85 found similar sodium excretions after sodium loads given intravenously or by gastric lavage. Therefore, the experimental evidence in favor of sodium or volume hepatic receptors remains controversial.
In summary, the afferent mechanisms for sodium and water retention in chronic heart failure may be preferentially localized on the arterial or high-pressure side of the circulation where EABV may serve as the primary determinant of the renal response. However, reflexes from the low-pressure cardiopulmonary receptor system also may be altered so as to influence renal sodium and water handling in heart failure. In this regard, increases in atrial and ventricular end-diastolic pressures also stimulate the release of the natriuretic peptides and inhibit AVP release, which may be important attenuating factors in renal sodium and water retention. Finally, these afferent mechanisms for initiating sodium and water retention in chronic heart failure should not be confused with additional mechanisms that may be implicated in the setting of acute decompensated heart failure, where increased central venous pressure and renal venous congestion may also contribute to worsening renal function and sodium and water retention, as discussed later in this chapter.
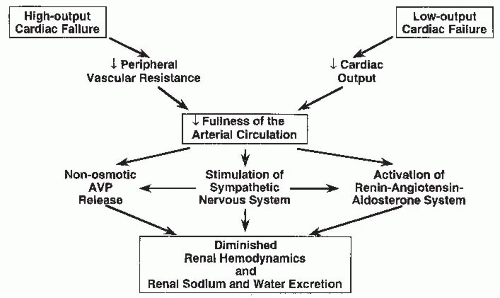 FIGURE 67.2 The mechanism explaining the defect in renal sodium and water excretion in both high- and low-output heart failure.AVP, arginine vasopressin. |
Efferent Mechanisms for Renal Sodium and Water Retention in Heart Failure
The Neurohormonal Response to Cardiac Failure
As mentioned, the activation of various neurohormonal vasoconstrictor and antinatriuretic systems mediates to a large extent the renal sodium and water retention associated with the edematous disorders. Arterial underfilling secondary to a diminished cardiac output or peripheral vasodilation, perhaps in association with an alteration in low-pressure baroreceptor function, elicits these “compensatory” neuroendocrine responses in order to maintain the integrity of the arterial circulation by promoting peripheral vasoconstriction and expansion of the ECF volume through renal sodium and water retention (Fig. 67.2). The three major neurohormonal vasoconstrictor systems activated in response to arterial underfilling are the sympathetic nervous system, the renin-angiotensin-aldosterone system, and the nonosmotic release of AVP. Although other vasoconstrictor hormones may also be activated in heart failure (e.g., endothelin), their role in heart failure pathophysiology remains unclear.
The baroreceptor activation of the sympathetic nervous system appears to be the primary integrator of the hormonal vasoconstrictor systems involved in renal sodium and water retention. The nonosmotic release of AVP involves sympathetic stimulation of the supraoptic and paraventricular nuclei in the hypothalamus,86 whereas activation of the renin-angiotensin-aldosterone system involves renal β-adrenergic stimulation.87 However, this latter system may provide positive feedback stimulation of the sympathetic nervous system and
nonosmotic AVP release. Various counterregulatory, vasodilatory, and natriuretic hormones, including the natriuretic peptides and PGs, are also activated in heart failure and the other edematous disorders, and may attenuate the renal effects of vasoconstrictor hormone activation. The effects of these neurohormonal systems, as well as the effects of alterations in systemic hemodynamics, on renal hemodynamics, and tubular sodium and water reabsorption in heart failure, are discussed in the following section.
nonosmotic AVP release. Various counterregulatory, vasodilatory, and natriuretic hormones, including the natriuretic peptides and PGs, are also activated in heart failure and the other edematous disorders, and may attenuate the renal effects of vasoconstrictor hormone activation. The effects of these neurohormonal systems, as well as the effects of alterations in systemic hemodynamics, on renal hemodynamics, and tubular sodium and water reabsorption in heart failure, are discussed in the following section.
Glomerular Filtration Rate
The GFR is usually normal in mild heart failure and is reduced only as cardiac performance becomes more severely impaired. Until 1961, it was generally accepted that the rate of glomerular filtration was a major determinant of renal sodium excretion. In 1961, de Wardener et al.88 published their classic paper indicating that acute expansion of ECF volume by saline loading was accompanied by a brisk natriuresis even when GFR was reduced. Moreover, in sodium-retaining heart failure patients, GFR is often normal and may even be elevated in states of high-output cardiac failure. These observations argue against an important role for diminished GFR in the sodium retention of heart failure per se (i.e., in the initiation of sodium retention), although a diminished GFR may be a contributing factor in patients with advanced heart failure or comorbid disorders that directly impair this aspect of renal function. It also should be emphasized that the contribution of GFR to sodium balance is difficult to evaluate because very minute changes in GFR are difficult to measure and may account for important changes in sodium excretion. For example, under normal conditions, with a GFR of 100 mL per minute, the filtered load of sodium amounts to approximately 20,000 mEq per day. This amount of filtered sodium is enormous compared to the normal urinary sodium excretion of approximately 200 mEq per day. In view of this considerable difference, it is apparent that very small changes in GFR can result in major alterations in sodium excretion if tubular reabsorption remains unaltered. In any event, although GFR may be diminished in patients with advanced heart failure, a reduction in GFR alone is probably not an important cause of fluid retention in these patients because sodium retention can be observed in heart failure patients who have GFRs comparable to normal subjects who are capable of maintaining sodium balance.
Renal Blood Flow
Heart failure is commonly associated with an increase in renal vascular resistance and a decrease in renal blood flow89 In general, renal blood flow decreases in proportion to the decrease in cardiac output. Some investigators also showed a redistribution of renal blood flow from the outer cortical nephron to juxtaglomerular nephrons during experimental heart failure.90,91 It was proposed that deeper nephrons with longer loops of Henle reabsorb sodium more avidly. Therefore, the redistribution of blood flow to these nephrons in patients with heart failure might account for or substantially contribute to the renal sodium retention observed. However, other investigators were not able to demonstrate such a redistribution of blood flow in other models of cardiac failure.92,93 At the present time, the role of redistribution of renal blood flow in the sodium retention of cardiac failure therefore remains uncertain.
The increased renal vascular resistance in heart failure could be caused by enhanced renal sympathetic activity or increased circulating concentrations of AT-II, norepinephrine, vasopressin, or other vasoconstricting substances. Alternatively, or in addition, decreased synthesis of or the development of tachyphylaxis to known vasodilating substances such as the natriuretic peptides and PGE2 and PGI2 may contribute to the increased renal vascular resistance. Studies performed in rats demonstrated the ability of the adrenergic neurotransmitter norepinephrine and AT-II to promote glomerular arteriolar constriction.94,95 In a rat model of low-output heart failure caused by myocardial infarction, the marked elevation in efferent arteriolar resistance was abolished after the infusion of an ACE inhibitor,95 thereby implicating the renal vasoconstrictor properties of AT-II in heart failure. Clinical results from our laboratory also favor AT-II as a major renal vasoconstrictive substance in patients with heart failure.96 In patients with advanced heart failure, GFR was improved after 1 month of treatment with the ACE inhibitor captopril. However, similar patients receiving another vasodilating agent, prazosin, with identical improvement in cardiac output and left ventricular end-diastolic pressure but without any effect on the renin-angiotensin system had no improvement in GFR.96 Moreover, a published review of the literature on renal function alterations induced by ACE inhibition during heart failure concluded that the net effect of ACE inhibitors in patients with heart failure is to augment renal blood flow to a greater extent than cardiac output.97 This observation also supports an important role for AT-II in the renal hemodynamic alterations of heart failure. However, the renal response to ACE inhibition in patients with heart failure is variable; as a result, it is acknowledged that volume status and the degree of neurohormonal activation may influence this response (see the following).
In heart failure, the interaction between norepinephrine or AT-II and PGs may also provide a means of preserving near constancy of renal blood flow in response to arterial underfilling. Although the inhibition of PG synthesis does not generally impair GFR in normovolemic animals98,99 or humans,100 in states of high plasma concentrations of endogenous AT-II induced by volume depletion, the blockade of PG synthesis may be associated with substantial declines in renal blood flow and GFR.98,99 Recent clinical results have underscored the importance of PGs in the maintenance of renal function in patients with heart failure.77,101 In patients with heart failure, PG activity is increased and correlates with the severity of disease as assessed by the degree of hyponatremia.77 In these 15 patients, plasma levels of the
metabolites of vasodilator PGI2 and PGE2 were found to be elevated 3 to 10 times above those seen in normal subjects. Of note, plasma levels of both metabolites also correlated positively with PRA and plasma AT-II concentrations. The administration of the PG synthesis inhibitor indomethacin in three of the hyponatremic heart failure patients resulted in a marked increase in peripheral vascular resistance and a fall in cardiac output. Riegger et al.101 recently evaluated the renal effects of another PG synthesis inhibitor, acetylsalicylic acid, in patients with moderate heart failure consuming a normal sodium diet. In these patients, acetylsalicylic acid in doses that decreased the synthesis of renal PGE2 resulted in a significant reduction in urinary sodium excretion. Moreover, the administration of a cyclooxygenase inhibitor in heart failure patients occasionally may result in acute reversible renal failure, an effect proposed to be due in part to the inhibition of vasodilating renal PGs and the resultant renal vasoconstriction.102 It should be noted, however, that the extent to which the effects on renal function and sodium and water handling result from renal hemodynamic or the tubular actions of the PGs remains unclear.
metabolites of vasodilator PGI2 and PGE2 were found to be elevated 3 to 10 times above those seen in normal subjects. Of note, plasma levels of both metabolites also correlated positively with PRA and plasma AT-II concentrations. The administration of the PG synthesis inhibitor indomethacin in three of the hyponatremic heart failure patients resulted in a marked increase in peripheral vascular resistance and a fall in cardiac output. Riegger et al.101 recently evaluated the renal effects of another PG synthesis inhibitor, acetylsalicylic acid, in patients with moderate heart failure consuming a normal sodium diet. In these patients, acetylsalicylic acid in doses that decreased the synthesis of renal PGE2 resulted in a significant reduction in urinary sodium excretion. Moreover, the administration of a cyclooxygenase inhibitor in heart failure patients occasionally may result in acute reversible renal failure, an effect proposed to be due in part to the inhibition of vasodilating renal PGs and the resultant renal vasoconstriction.102 It should be noted, however, that the extent to which the effects on renal function and sodium and water handling result from renal hemodynamic or the tubular actions of the PGs remains unclear.
As mentioned, norepinephrine may also contribute to the increased renal afferent arteriolar resistance in heart failure patients. In this regard, Oliver et al.103 demonstrated that the venous to arterial norepinephrine concentration gradient across the kidney, a crude measure of renal nerve traffic, is increased in response to acute reduction of cardiac output. Moreover, Hasking et al.104 showed that during a steady-state tritiated norepinephrine infusion, the spillover of norepinephrine to plasma from the kidney is significantly elevated in patients with heart failure. In these patients, the increased renal norepinephrine spillover substantially contributed to the increase in whole-body norepinephrine spillover. These findings demonstrate that renal adrenergic activity is increased in patients with heart failure, and thus contributes to the renal vasoconstriction. In support of this latter hypothesis, the administration of α-adrenergic receptor antagonists increased renal blood flow in edematous patients with heart failure.105 Renal denervation studies in patients with refractory hypertension also underscore the role of renal sympathetic nerve activation in cardiovascular disease. In such patients, the catheter ablation of renal nerves reduces norepinephrine spillover from the kidneys and lowers blood pressure.106,107,108 Ongoing renal denervation studies in patients with heart failure may shed further light on the role of renal sympathetic activation and the potential for catheter ablation of renal nerves in the treatment of heart failure.
Filtration Fraction, Proximal Tubular Sodium and Water Reabsorption, and Factors Acting Beyond the Proximal Tubule
Because renal blood flow falls as cardiac output decreases and GFR is usually preserved, the filtration fraction often is increased in early heart failure. An increase in the filtration fraction results in increased protein concentration and oncotic pressure in the efferent arterioles and the peritubular capillaries that surround the proximal tubule.95 Such an increase in peritubular oncotic pressure has been proposed to increase sodium and water reabsorption in the proximal tubule.109,110,111,112,113 Direct evidence for increased single-nephron filtration fraction was provided by micropuncture studies in rats with myocardial infarction induced by coronary ligation.95 In rats with large myocardial infarctions involving approximately 40% of the left ventricular circumference, the single-nephron filtration fraction was markedly elevated (0.38 ± 0.02 versus 0.25 ± 0.02, P < .005) when compared with that in sham-operated control rats. The measurement of preglomerular, glomerular, and postglomerular pressures and flows revealed that these reductions in glomerular plasma flow rate and elevations in filtration fraction were associated with a profound constriction of the efferent arterioles. The effect of the latter was to sustain glomerular capillary hydraulic pressure, thereby preventing a marked fall in GFR. Significantly, fractional proximal fluid reabsorption was elevated in this model. Of interest, in these animals with myocardial infarction, the intravenous infusion of the ACE inhibitor teprotide led to the return of glomerular plasma flow rate, single-nephron filtration fraction, single-nephron GFR, efferent arteriolar resistance, and fractional proximal fluid reabsorption to or toward the levels found in the control rats.95 Consistent with these experiments, micropuncture studies performed in other models of heart failure such as acute TIVC constriction114 and acute cardiac tamponade115 in dogs showed that the proximal tubule was at least one major nephron site responsible for renal sodium retention or a blunted response to saline infusion.
Despite the convincing nature of many studies, not all investigators have been able to detect an effect of peritubular oncotic pressure on proximal tubular sodium and water reabsorption. Rumrich and Ullrich,116 Lowitz et al.,117 Bank et al.,118 and Holzgreve and Schrier119 were unable to find changes in proximal reabsorption despite marked changes in peritubular oncotic pressures. Moreover, Conger et al.120 directly perfused peritubular capillaries with either a protein-free or protein-rich solution and found that neither perfusate influences the rate of proximal reabsorption. Trying to reconcile these observations, Ott et al.121 found that proximal reabsorption was different after changes in peritubular oncotic pressure in volume-expanded dogs compared with hydropenic animals. These authors suggested that the expansion of ECF volume resulted in an increased passive back leak that could be reversed by raising the peritubular oncotic pressure. During hydropenia, however, when passive back leak was relatively less, raising the peritubular capillary oncotic pressure did not influence proximal reabsorption.
The effects of increased filtration fraction might be expected to be exerted primarily on proximal tubular sodium reabsorption. Nevertheless, although clearance and micropuncture studies in animals with heart failure have demonstrated increased sodium reabsorption in the proximal tubule, distal
sodium reabsorption also seems to be involved. In this regard, clearance and micropuncture studies performed in dogs with AV fistulae,122 chronic pericarditis,115 and chronic partial thoracic vena caval obstruction123 documented enhanced distal nephron sodium reabsorption. Levy123 also showed that the inability of dogs with chronic vena caval obstruction to excrete a sodium load is a consequence of enhanced reabsorption of sodium at the loop of Henle. This nephron segment was similarly implicated in rats with AV fistulae.93 Physical factors also could be involved in the augmented reabsorption of sodium chloride by the loop of Henle in dogs with constriction of the vena cava.123
sodium reabsorption also seems to be involved. In this regard, clearance and micropuncture studies performed in dogs with AV fistulae,122 chronic pericarditis,115 and chronic partial thoracic vena caval obstruction123 documented enhanced distal nephron sodium reabsorption. Levy123 also showed that the inability of dogs with chronic vena caval obstruction to excrete a sodium load is a consequence of enhanced reabsorption of sodium at the loop of Henle. This nephron segment was similarly implicated in rats with AV fistulae.93 Physical factors also could be involved in the augmented reabsorption of sodium chloride by the loop of Henle in dogs with constriction of the vena cava.123
Intrarenal mechanisms, specifically decreased delivery of tubular fluid to the distal diluting segment of the nephron, may also contribute to the impaired water excretion observed in heart failure. Evidence supporting this intrarenal mechanism of water retention in heart failure has been provided by studies involving the administration of mannitol124 or the loop diuretic furosemide125 to patients with heart failure and hyponatremia. The administration of either of these agents converted the cardiac patient’s hypertonic urine to a dilute urine.124,125 Both mannitol and furosemide may diminish the tubular reabsorption of sodium and water in the more proximal portions of the nephron, thus increasing fluid delivery to the more distal nephron sites of urinary dilution. Other factors may, however, be implicated to explain these results: (1) the infusion of mannitol may produce volume expansion, thereby suppressing the baroreceptor-mediated release of AVP; and (2) the furosemide-induced hypotonic urine was found to not be responsive to the administration of exogenous AVP, thus suggesting antagonism of AVP by furosemide.125 In support of this latter hypothesis, Szatalowicz et al.126 provided further evidence that furosemide interferes with the renal action of AVP in humans.
In summary, the exact contribution of proximal versus distal nephron sites in the augmented sodium and water reabsorption seen in heart failure may depend on the severity of the heart failure and the concomitant degree of arterial underfilling. The fact that changes in the filtration fraction have been observed in patients with heart failure before changes in sodium balance occur may question the dominance of peritubular factors and proximal reabsorption in the sodium retention characteristic of heart failure.127 This observation suggests that other factors, such as the direct tubular effects of neurohormonal activation, may play a significant role in the renal sodium and water retention of heart failure. The renal effects of these various neurohormonal systems are discussed in detail in the following section, starting with activation of the vasoconstrictor mechanisms.
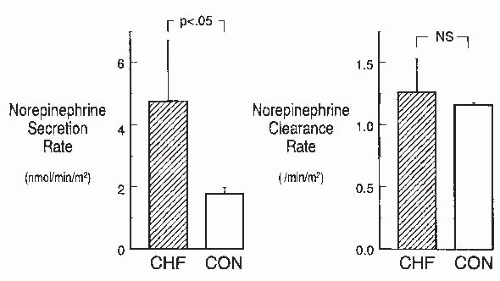 FIGURE 67.3 Plasma norepinephrine secretion and clearance rates in patients with mild-to-moderate heart failure (CHF) and in normal control subjects (CON). The findings of increased norepinephrine secretion and normal norepinephrine clearance in the CHF patients are consistent with early activation of the sympathetic nervous system in cardiac failure. NS, not significant. (From Abraham WT, Hensen J, Schrier RW. Elevated plasma noradrenaline concentrations in patients with low-output cardiac failure: dependence on increased noradrenaline secretion rates. Clin Sci. 1990;79:429, with permission.) |
Vasoconstrictor Systems
Activation of the sympathetic nervous system in heart failure. The sympathetic nervous system is activated early in patients with heart failure. Numerous studies have documented elevated peripheral venous plasma norepinephrine concentrations in heart failure patients.104,128,129,130,131 In advanced heart failure, using tritiated norepinephrine to determine norepinephrine kinetics, Hasking et al.104 and Davis et al.130 demonstrated that both increased norepinephrine spillover and decreased norepinephrine clearance contribute to the elevated venous plasma norepinephrine levels seen in these patients, suggesting that increased sympathetic nerve activity is at least partially responsible for the high circulating norepinephrine levels. Our laboratory131 has demonstrated that in earlier stages of heart failure, the rise in plasma norepinephrine in patients with heart failure was due solely to increased norepinephrine secretion (Fig. 67.3), supporting the notion that sympathetic nervous system activity is increased early in the course of heart failure. Significantly, in our heart failure patients with mild-to-moderate symptoms, plasma epinephrine, a marker of adrenal activation, was not substantially elevated, confirming the neuronal source of the increased norepinephrine.
The Studies of Left Ventricular Dysfunction (SOLVD) investigators132 reported the presence of adrenergic activation in patients with asymptomatic left ventricular dysfunction. In this substudy of the SOLVD trials, neurohormonal activation was assessed in 56 control subjects, 151 patients with left ventricular dysfunction (ejection fractions ≤ 35%) but no overt heart failure, and 81 patients with overt heart failure, prior to randomization to receive placebo versus an ACE inhibitor. The plasma norepinephrine concentration
was significantly increased by 35% in subjects with asymptomatic left ventricular dysfunction compared to healthy control subjects, and by 65% greater than control values in the overt heart failure patients. These data also demonstrate that adrenergic activation occurs early in the course of heart failure or left ventricular dysfunction and are consistent with the observation that plasma norepinephrine concentrations or the degree of adrenergic activation are directly correlated with the degree of left ventricular dysfunction in patients with heart failure.128,129,133,134 Finally, studies employing peroneal nerve microneurography to directly assess sympathetic nerve activity to muscle (MSNA) confirmed increased adrenergic nerve traffic in patients with heart failure.135
was significantly increased by 35% in subjects with asymptomatic left ventricular dysfunction compared to healthy control subjects, and by 65% greater than control values in the overt heart failure patients. These data also demonstrate that adrenergic activation occurs early in the course of heart failure or left ventricular dysfunction and are consistent with the observation that plasma norepinephrine concentrations or the degree of adrenergic activation are directly correlated with the degree of left ventricular dysfunction in patients with heart failure.128,129,133,134 Finally, studies employing peroneal nerve microneurography to directly assess sympathetic nerve activity to muscle (MSNA) confirmed increased adrenergic nerve traffic in patients with heart failure.135
As mentioned, studies in human heart failure demonstrated the presence of renal adrenergic activation.104 In this study of whole-body and organ-specific norepinephrine kinetics in heart failure patients, cardiac and renal norepinephrine spillovers were increased 504% and 206%, respectively, whereas norepinephrine spillover from the lungs was normal. These findings demonstrate the presence of selective cardiorenal adrenergic activation in heart failure. A discussion of the cardiac effects of this adrenergic activation is beyond the scope of this chapter. However, low heart rate variability (indicative of high cardiac sympathetic and low cardiac parasympathetic activity) assessed continuously by implantable pacemaker and/or defibrillator devices is a predictor of hospitalization for worsening heart failure.136 Of note, in this report most hospitalizations for worsening heart failure were associated with fluid-volume overload. Therefore, measuring heart rate variability may provide insight into the systemic as well as the cardiac effects of heightened adrenergic activity. Moreover, numerous adverse effects of increased cardiac adrenergic activity have been documented in humans,137 and positive experience with the use of β-adrenergic receptor antagonists in heart failure patients137,138
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Hypertension due to Primary Aldosteronism and Related Disorders
Hypertension due to Primary Aldosteronism and Related Disorders
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis




Related posts:
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Pathophysiology of Nephrotoxic Cell Injury
Hypertension due to Primary Aldosteronism and Related Disorders
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
