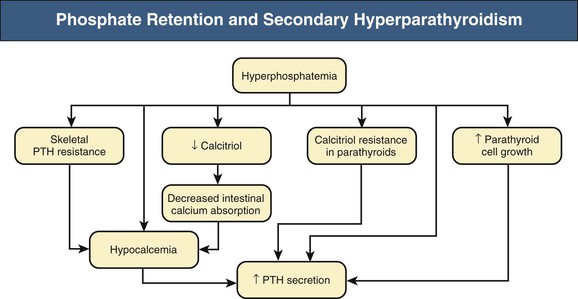
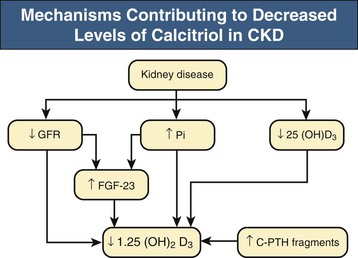
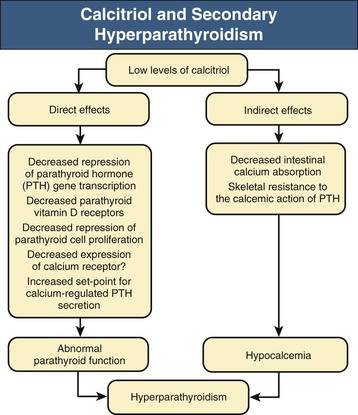
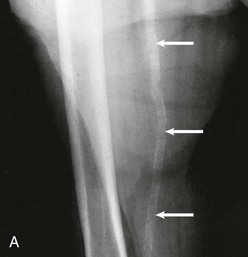
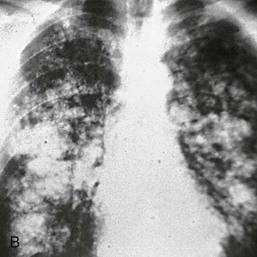
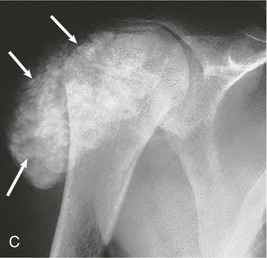
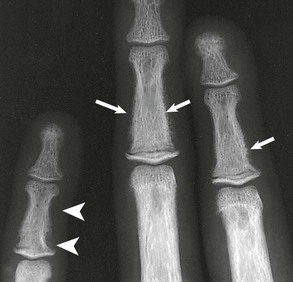
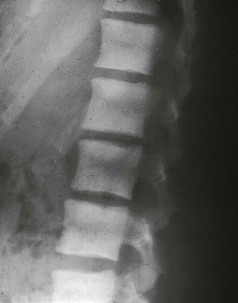
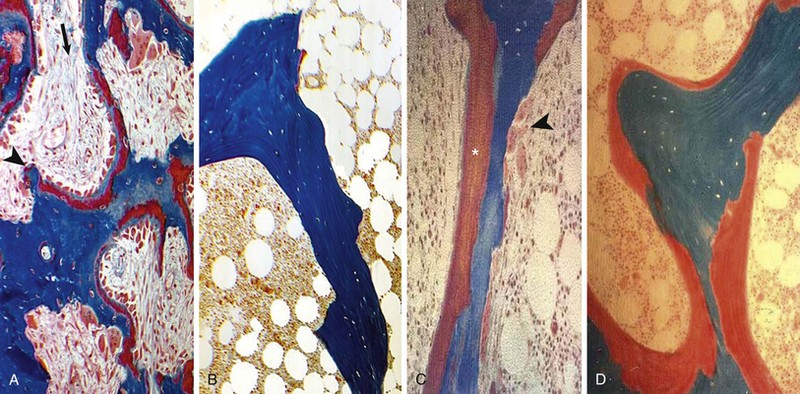
Kevin J. Martin, Jürgen Floege, Markus Ketteler Disturbances of mineral metabolism are common if not ubiquitous during the course of chronic kidney disease (CKD) and lead to serious and debilitating complications unless these abnormalities are addressed and treated. The spectrum of disorders includes abnormal concentrations of serum calcium, phosphate, and magnesium and disorders of parathyroid hormone (PTH), fibroblast growth factor 23 (FGF-23), and vitamin D metabolism. These abnormalities as well as other factors related to the uremic state affect the skeleton and result in the complex disorders of bone known as renal osteodystrophy; it is now recommended that this term be used exclusively to define the bone disease associated with CKD. The clinical, biochemical, and imaging abnormalities heretofore identified as correlates of renal osteodystrophy should be defined more broadly as a clinical entity or syndrome called chronic kidney disease–mineral and bone disorder (CKD-MBD).1 The spectrum of skeletal abnormalities seen in renal osteodystrophy includes the following (Fig. 85-1): ▪ Adynamic bone disease (ABD), a condition characterized by abnormally low bone turnover. ▪ Combinations of these abnormalities termed mixed renal osteodystrophy. The prevalence of the various types of renal bone disease in patients with end-stage renal disease (ESRD) is illustrated in Figure 85-2.2 In patients on hemodialysis, osteitis fibrosa and ABD now occur with almost equal frequency. In contrast, in patients on peritoneal dialysis, the adynamic bone lesion predominates. Osteomalacia represents only a small fraction of cases in either group but is more common in certain ethnic groups, particularly Indo-Asians. The abnormalities of the skeleton start relatively early in the course of CKD. Several biochemical and hormonal abnormalities that are encountered during the course of CKD contribute to renal bone disease and can be affected by efforts at prevention and therapy. The major factors that are operative in early CKD may vary as CKD progresses (Fig. 85-3). Similarly, the predominance of one particular pathogenetic mechanism over another may contribute to the heterogeneity of bone disorders. We therefore discuss separately the two major entities, namely, high- and low-turnover osteodystrophy. Elevated levels of PTH in blood, hyperplasia of the parathyroid glands, and elevations in FGF-23 are seen early in CKD. Whereas the level of free (i.e., non–protein bound) calcium in blood is normally the principal determinant of PTH secretion, during the course of CKD, several metabolic disturbances also alter the regulation of the secretion of PTH. There are three main body pools of calcium: the bony skeleton (mineral component), the intracellular pool (mostly protein bound), and the extracellular pool (see Chapter 10). The calcium in the extracellular pool is in continuous exchange with that of bone and cells and is altered by diet and excretion. Calcium metabolism depends on the close interaction of two hormonal systems: PTH and vitamin D. Perturbations of both of these systems occur during the course of CKD, with adverse consequences on the skeleton. Total serum calcium tends to decrease during the course of CKD as a result of phosphate retention and decreased production of 1,25-dihydroxyvitamin D (calcitriol) from the kidney, decreased intestinal calcium absorption, and skeletal resistance to the calcemic action of PTH, but the levels of free calcium remain within the normal range in most patients3 as a result of compensatory hyperparathyroidism. Because calcium is a major regulator of PTH secretion, persistent hypocalcemia is a powerful stimulus for the development of hyperparathyroidism and also contributes to parathyroid growth. With progressive CKD, phosphate is retained, at least, transiently, by the failing kidney. However, hyperphosphatemia usually does not become evident before CKD stage 4. Until then, compensatory hyperparathyroidism and increases in circulating FGF-23 result in increased phosphaturia, maintaining serum phosphate levels in the normal range.4 One mechanism by which phosphate retention may lead to hyperparathyroidism is by a decrease in serum free calcium, which in turn stimulates the secretion of PTH (Fig. 85-4). Thus, a new steady state is achieved in which serum phosphate is restored to normal at the expense of a sustained high level of PTH. This cycle is repeated as renal function declines until sustained and severe hyperparathyroidism is present. Second, phosphate retention leads to decreased production of calcitriol by the kidney, either directly or by increasing the levels of FGF-23 (which decreases the activity of 1α-hydroxylase). The decrease in calcitriol allows increases in PTH gene transcription by direct action and also decreases intestinal calcium absorption, leading to hypocalcemia, which in turn stimulates PTH secretion. Third, hyperphosphatemia is associated with resistance to the actions of calcitriol in the parathyroid glands, which also favors the development of hyperparathyroidism and also induces resistance to the actions of PTH in bone. Finally, phosphate per se appears to affect PTH secretion independently of changes in serum calcium or serum calcitriol.5,6 Phosphate may also have an effect on parathyroid growth independent of serum calcium.7,8 Regardless of the mechanism by which phosphate retention causes hyperparathyroidism, restriction of dietary phosphate in proportion to the decrease in glomerular filtration rate (GFR), in experimental animals, can prevent the development of hyperparathyroidism. Current evidence suggests that FGF-23 also acts directly on the parathyroid gland and has inhibitory effects on PTH secretion and parathyroid growth.9,10 This would suggest that the main effects of FGF-23 on the pathogenesis of hyperparathyroidism appear to be indirect as a result of the potent effect of FGF-23 to decrease calcitriol production. These various actions may explain the association of the levels of FGF-23 with patient outcome.11 The conversion of 25-hydroxyvitamin D to its active metabolite 1,25-dihydroxyvitamin D occurs mainly in the kidney by the enzyme 1α-hydroxylase. Extrarenal production of calcitriol also occurs and contributes to the circulating levels of calcitriol. Renal calcitriol production progressively declines during the course of CKD as a result of several mechanisms (Fig. 85-5). Calcitriol production is compromised in the setting of CKD by a reduction in 25-hydroxyvitamin D levels12 and the decrease in GFR, which further limits the delivery of 25-hydroxyvitamin D to the site of the 1α-hydroxylase in the proximal tubule. Phosphate retention either directly or by inducing an increase in FGF-23 also decreases the activity of 1α-hydroxylase. Finally, it appears that circulating PTH fragments may also directly decrease calcitriol production. The resultant decreased levels of calcitriol contribute to the pathogenesis of hyperparathyroidism by several direct and indirect mechanisms (Fig. 85-6). Low levels of calcitriol directly release the gene for PTH from suppression by the vitamin D receptor and allow increased PTH secretion. In many tissues, vitamin D regulates its own receptor by positive feedback; the vitamin D receptor content is decreased in parathyroid tissue in CKD. Administration of calcitriol has been shown to increase the vitamin D receptor content in the parathyroid glands coincident with the suppression of PTH secretion. Studies in vitro have shown that calcitriol is a negative growth regulator of parathyroid cells; therefore, calcitriol deficiency in patients with CKD may facilitate parathyroid cell proliferation. Other direct consequences of low levels of calcitriol that contribute to the pathogenesis of secondary hyperparathyroidism include an increase in the parathyroid set-point for calcium-regulated PTH secretion and possibly a decrease in the expression of calcium receptors. Low levels of calcitriol may also promote the development of hyperparathyroidism indirectly. First, decreased calcitriol production as renal function decreases can lead to progressive reductions in intestinal absorption of calcium, leading to hypocalcemia and stimulation of PTH release. Second, low levels of calcitriol have been implicated in skeletal resistance to the calcemic actions of PTH, which may also contribute to the development of hyperparathyroidism. There are intrinsic abnormalities in parathyroid gland function in the course of CKD in addition to those caused by hypocalcemia, low levels of calcitriol, and skeletal resistance to the actions of PTH (Box 85-1). Parathyroid hyperplasia is an early finding in CKD. In experimental models, hyperplasia begins within a few days after the induction of CKD and can be prevented by dietary phosphate restriction or by the use of calcimimetic agents.7,13 Resected parathyroids from patients with severe hyperparathyroidism have nodular areas throughout the gland, which represent monoclonal expansions of parathyroid cells.14 Within these nodules, there is decreased expression of vitamin D receptors as well as of calcium receptors.15,16 The decreased expression of calcium receptors renders efforts to therapeutically affect these enlarged hyperplastic glands difficult. The parathyroid calcium receptor is centrally involved in the regulation of PTH secretion by calcium.17 Its expression and synthesis are decreased in parathyroid glands from hyperparathyroid patients,16 leading to altered calcium-regulated PTH secretion. Increased concentrations of calcium are required in vitro to suppress PTH release from the parathyroid cells of uremic patients compared with those of normal controls. Thus, the set-point for the concentration of calcium required to decrease PTH release by 50% appears to be increased. In patients with CKD, there is an impaired response of serum calcium to the administration of PTH and a delay in the recovery from induced hypocalcemia in the presence of larger increments in PTH levels. Thus, in CKD the skeleton is relatively resistant to the calcemic actions of PTH. The resultant decrease in serum calcium levels stimulates PTH secretion and contributes to the pathogenesis of secondary hyperparathyroidism. Factors involved in the skeletal resistance to PTH in CKD include decreased levels of calcitriol, downregulation of the PTH receptor, and phosphate retention. In addition, circulating fragments of PTH, truncated at the N-terminus, which still react in the older, second-generation two-site PTH assays, may serve to oppose the calcemic actions of PTH, probably acting at a receptor for the C-terminal region of PTH.18,19 Clinical manifestations of hyperparathyroidism are usually nonspecific and are often preceded by biochemical or imaging abnormalities. Aches and pains are common manifestations, often nonspecific in nature; occur in the lower back, hips, and legs; and are aggravated by weight bearing. Acute, localized bone pain can also manifest and may be suggestive of acute arthritis. Pain around joints may be caused by acute periarthritis, which is associated with periarticular deposition of calcium phosphate crystals, especially in patients with marked hyperphosphatemia. The symptoms may be confused clinically with gout or pseudogout and often respond to nonsteroidal anti-inflammatory drugs (NSAIDs). The gradual onset of muscle weakness is also common in patients with ESRD. Many factors are probably involved in its pathogenesis, including hyperparathyroidism and abnormalities of vitamin D. The arthropathy associated with β2-microglobulin amyloidosis (see later discussion) should be considered in the differential diagnosis in very-long-term dialysis patients. Bone deformities may occur in patients with severe hyperparathyroidism, particularly in children. In adults, deformities arise as a consequence of fractures, sometimes induced by brown tumors (see later discussion); the axial skeleton is most commonly affected. This can lead to kyphoscoliosis or chest wall deformities. Slipped epiphysis may occur in children, and frank rachitic features are occasionally evident. Growth retardation is also common in children, and although some improvement has been shown with calcitriol, this has not been the universal finding. Extraskeletal calcifications are frequently encountered in patients with advanced CKD and are aggravated by persistent elevation of the calcium-phosphate product. Most commonly, vascular calcifications are seen, but calcifications may also occur in other sites, such as the lung, myocardium, and periarticular areas (see Fig. 85-7). In the skin, hyperparathyroidism can manifest as pruritus (discussed in detail in Chapter 88). Rarely, it can also underlie the development of calciphylaxis, or calcific uremic arteriolopathy (discussed in detail in Chapter 88; see Figs. 88-6 and 88-7). In addition to the clinical manifestations of renal osteodystrophy, a variety of biochemical and radiographic techniques are helpful to establish the specific diagnosis and to serve as a guide for the initiation and adjustment of therapeutic interventions. Although bone biopsy is not widely used in clinical practice, it remains the gold standard for the diagnosis of renal osteodystrophy. The levels of free calcium and phosphate in serum are usually normal in patients with mild to moderate CKD. Usually in stage 4 CKD, the levels of serum calcium tend to fall and hyperphosphatemia manifests. Hypercalcemia may result from the administration of large doses of calcium-containing antacids or the administration of vitamin D metabolites. Patients with severe hyperparathyroid bone disease may develop hypercalcemia. It is important to differentiate among the different causes of hypercalcemia in the setting of CKD (see Chapter 10) because the management will vary greatly according to the cause. Also, the levels of serum calcium and phosphate, when used alone, are not useful in predicting the specific type of bone disease. Measurements of PTH are important for diagnostic purposes and for therapeutic guidance in the management of renal osteodystrophy. With renal impairment, there is accumulation of circulating PTH fragments, which complicates the interpretation of PTH assays, including the two-site immunometric assays, which were thought to measure “intact” PTH (iPTH). Refinements in PTH assays have demonstrated that these iPTH assays also measure some large fragments of PTH, which are truncated at the N-terminus and may have important biologic activity. Although more specific assays for so-called “biointact” PTH (PTH 1-84) have been developed that exclude these fragments from measurement, work continues to define the clinical usefulness of such more specific assays.20–22 Recent demonstration of the presence of oxidized PTH, which is biologically inactive, in patients with ESRD may further complicate PTH measurements.23 It is hoped that continued efforts will lead to improved standardization among different PTH assays from various laboratories and various manufacturers of assay reagents. With existing iPTH assays (upper limit of the reference range, approximately 60 pg/ml), only values at the extremes are useful in the noninvasive diagnosis of renal osteodystrophy. In dialysis patients, iPTH levels above approximately 600 pg/ml are characteristic of patients with osteitis fibrosa. It is well accepted that there is an element of skeletal resistance to PTH in patients with CKD; therefore supranormal levels of PTH appear to be required to maintain normal bone turnover. Serial measurements of PTH are useful in the initial evaluation of patients with renal bone disease and are essential during the management of these disorders to assess response to therapy and to avoid overtreatment and undertreatment because either can have detrimental effects on bone histology. There are marked differences between commercial PTH assay results so that precise recommendations of desired ranges cannot reliably be provided.24 The levels of calcitriol in patients with CKD are not helpful in differentiating the histologic lesions of renal osteodystrophy. Measurements of calcitriol are not used routinely for diagnostic purposes unless extrarenal production of this metabolite is suspected, as in granulomatous disorders (see Chapter 10). Vitamin D deficiency in CKD rarely results in osteomalacia in the United States and Europe but may contribute to hyperparathyroidism. In patients with CKD with proteinuria, there is loss of vitamin D–binding protein in the urine, which may result in decreased levels of 25-hydroxyvitamin D. Vitamin D deficiency may be encountered in patients with limited sun exposure, in those with intestinal malabsorption or malnutrition, and in susceptible racial groups, particularly Indo-Asians. Assessment of vitamin D nutrition is by measurement of serum 25-hydroxyvitamin D3. Levels of circulating alkaline phosphatase offer an approximate index of osteoblast activity in patients with CKD. High levels are commonly present in hyperparathyroid bone disease. The discriminatory power of alkaline phosphatase measurements is enhanced by measurement of bone-specific alkaline phosphatase (BAP) isoenzyme and in particular in conjunction with PTH values. Serial measurements of alkaline phosphatase may be useful in assessing the progression of bone disease. Osteocalcin is another marker of osteoblastic activity, but it is not superior to alkaline phosphatase. Tartrate-resistant acid phosphatase and collagen degradation products are both markers of osteoclastic activity but are considered investigational at present. Routine x-ray examination of the skeleton is relatively insensitive for the diagnosis of renal osteodystrophy, and radiographs can appear virtually normal in patients with severe histologic evidence of renal bone disease. However, subperiosteal erosions are often present in severe secondary hyperparathyroidism, detected in the hands (Fig. 85-8), clavicles, and pelvis. Skull radiographs may show focal radiolucencies and a ground-glass appearance, known as “pepper pot” skull. Osteosclerosis of the vertebrae is responsible for the “rugger-jersey” appearance of the spine (Fig. 85-9). Very rarely, brown tumors, focal collections of giant cells and typical of severe hyperparathyroidism, are seen as well-demarcated radiolucent zones in long bones, clavicles, and digits. They may be confused with osteolytic metastases. Looser zones or pseudofractures are characteristic of osteomalacia. Routine skeletal radiographs are not indicated unless there are symptoms. Dual-energy x-ray absorptiometry is widely used to assess bone density. However, there is no clear usefulness for this technique in the assessment of renal bone disease (see later discussion) because bone density measurements do not correlate with bone histology in renal bone disease. Vascular and soft tissue calcifications may contribute to errors in bone density measurements. Biopsy of bone and the microscopic analysis of undercalcified sections after double tetracycline labeling provide definitive and quantitative diagnosis of renal bone disease.25 To standardize reports on bone histology, the Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD work group proposed the TMV classification, an assessment of turnover (T), mineralization (M), and bone volume (V).1 Bone mineralization is assessed by the administration of two different tetracyclines spaced apart (e.g., tetracycline 500 mg three times daily for 2 days, followed by a 10-day interval, then demeclocycline 300 mg three times daily for 3 days) and biopsy 4 days later; the quantitation of bone mineralization rate is achieved by measuring the distance between the two fluorescent tetracycline bands. Bone biopsy is not routinely performed in clinical practice because of the invasive nature of the procedure. Although noninvasive testing is useful to distinguish normal from high bone turnover, there is considerable overlap, and therefore biopsy might be required for definitive diagnosis when biochemistry is not conclusive (e.g., PTH in recommended range but bone alkaline phosphatase elevated, hypercalcemia with PTH only modestly elevated, or bone pain). Osteitis fibrosa (hyperparathyroid bone disease) is characterized by increased bone turnover, increased number and activity of osteoblasts and osteoclasts, and variable amounts of peritrabecular fibrosis (Fig. 85-10, A). Osteoid may be increased but usually has a woven pattern distinct from the normal lamellar appearance. Osteomalacia is characterized by increased osteoid seam width, increase in the trabecular surface covered with osteoid, and decreased bone mineralization as assessed by tetracycline labeling (Fig. 85-10, B). The presence of aluminum can be detected on the mineralization front by specific staining (Fig. 85-10, C). Aluminum-related bone disease is defined by aluminum staining exceeding 15% of the trabecular surface and a bone formation rate of less than 220 mm2/day. Features of osteitis fibrosa may occur together with features of osteomalacia; the combination is termed mixed renal osteodystrophy.
Bone and Mineral Metabolism in Chronic Kidney Disease
Definition
Epidemiology
Pathogenesis
Osteitis Fibrosa: Hyperparathyroidism—High-Turnover Renal Bone Disease
Abnormalities of Calcium Metabolism
Abnormalities of Phosphate Metabolism
Abnormalities of Vitamin D Metabolism
Abnormalities of Parathyroid Gland Function
Abnormal Skeletal Response to Parathyroid Hormone
Clinical Manifestations of High-Turnover Renal Osteodystrophy
Diagnosis and Differential Diagnosis
Serum Biochemistry
Parathyroid Hormone
Vitamin D Metabolites
Markers of Bone Formation and Bone Resorption
Radiology of the Skeleton
Measurements of Bone Density
Bone Biopsy
Treatment of High-Turnover Bone Disease
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Bone and Mineral Metabolism in Chronic Kidney Disease
Chapter 85
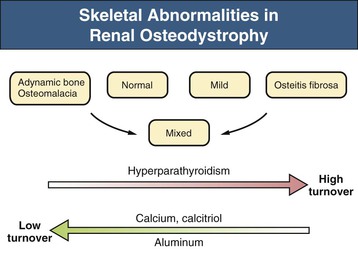
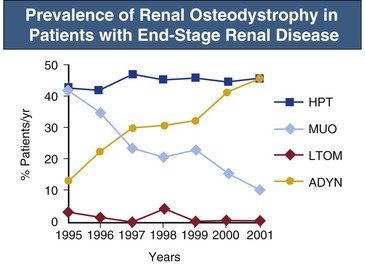
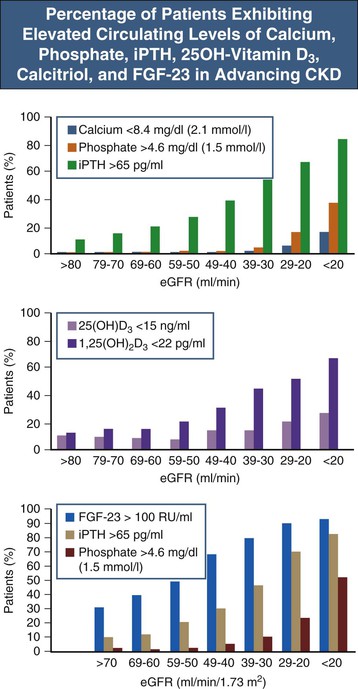
Figure 85-3 Percentage of patients exhibiting elevated circulating levels of calcium, phosphate, iPTH, 25OH-vitamin D3, calcitriol, and FGF-23 in advancing CKD. In particular in early CKD stages there is wide variability at the individual level, and some patients, for example, may exhibit increased FGF-23 and normal iPTH, whereas others may have elevated iPTH levels with normal FGF-23 or elevations of both. FGF-23, Fibroblast growth factor 23; iPTH, intact PTH. (Data from references 65 and 66.)