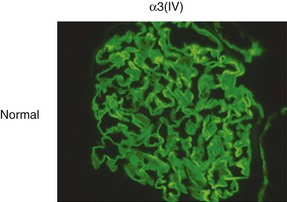
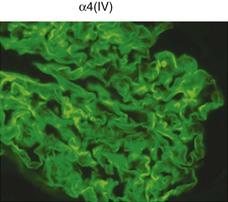
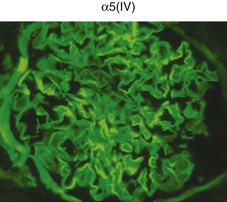
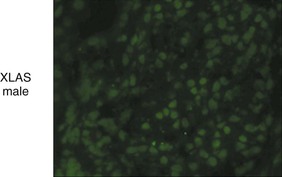
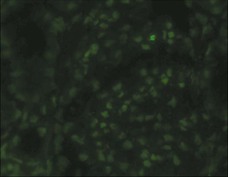
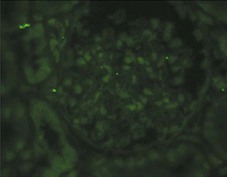
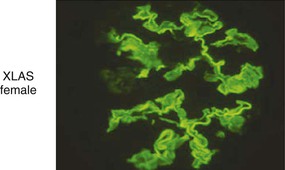
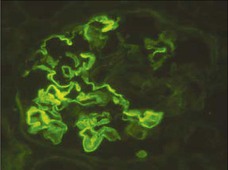
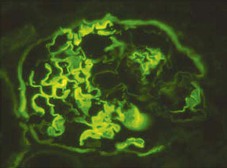
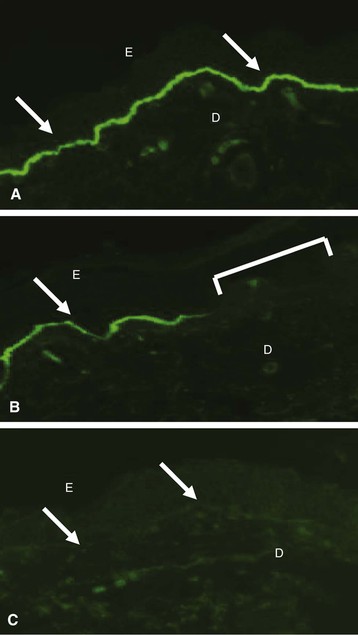
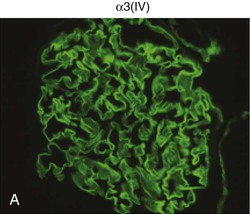
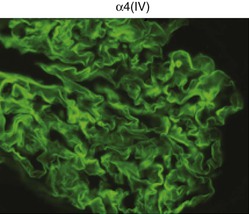
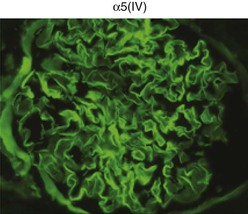
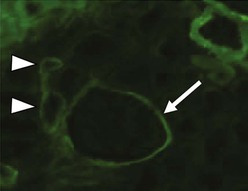
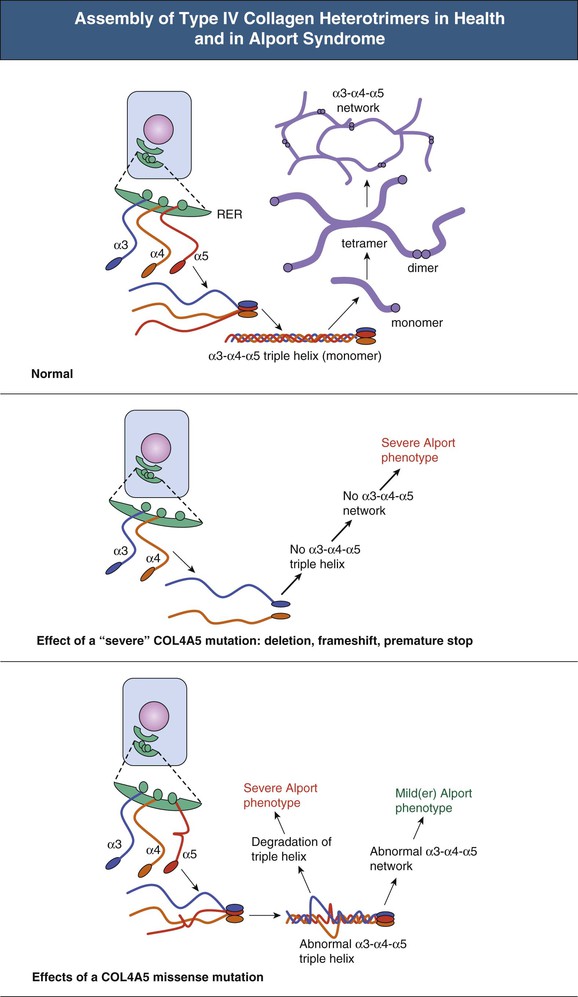
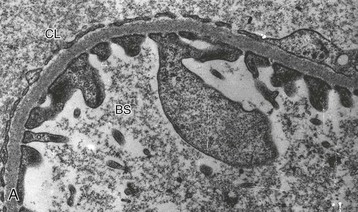
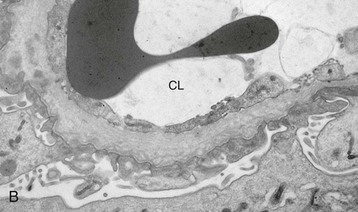
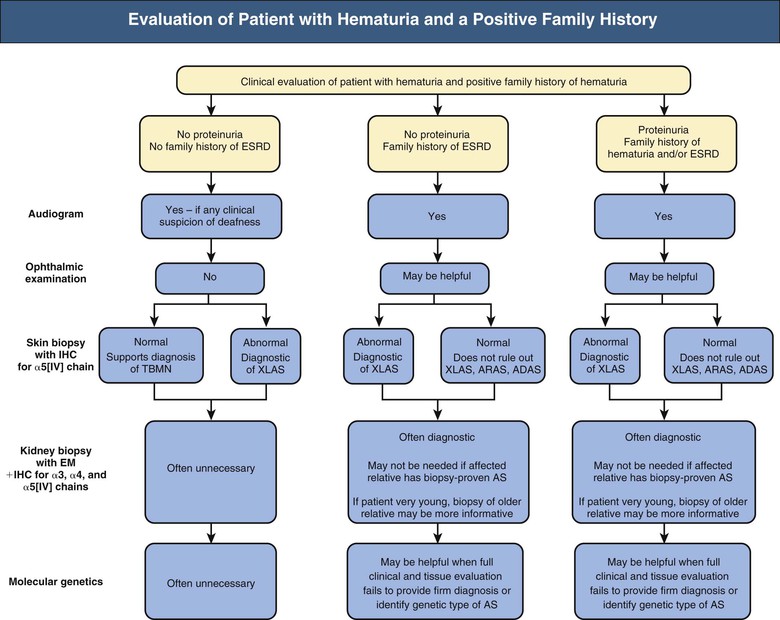
Clifford E. Kashtan Alport syndrome (AS) is a generalized, inherited disorder of basement membranes caused by mutations affecting specific proteins of the type IV (basement membrane) collagen family. The major features of AS are hematuria, progressive nephritis with proteinuria and declining renal function, sensorineural deafness, and ocular abnormalities. The course of AS is gender dependent; affected males typically have severe disease, whereas the manifestations of AS in women tend to be relatively mild. In 1902, Guthrie1 provided the first description of familial hematuria. Later studies of Guthrie’s family by Hurst2 and Alport3 revealed the progressive nature of the nephropathy, its association with deafness, and the poorer prognosis in affected males. In the 1970s, the glomerular basement membrane (GBM) was recognized as the site of the primary abnormality in AS.4–6 Indirect evidence of abnormalities in type IV collagen7,8 was followed by mapping of the major AS locus to the X chromosome,9 cloning of a new type IV collagen gene (COL4A5) and its assignment to the same X-chromosomal region,10 and identification of the first COL4A5 mutations in patients with X-linked AS.11 Type IV collagen is a major constituent of basement membranes. The type IV collagen family of proteins comprises six isomeric chains, designated α1(IV) to α6(IV). Each of these chains has a major collagenous domain of about 1400 residues containing the repetitive triplet sequence glycine (Gly)–X–Y, in which X and Y represent a variety of other amino acids; a C-terminal noncollagenous (NC1) domain of about 230 residues; and a noncollagenous N-terminal sequence of 15 to 20 residues. Each type IV collagen molecule is a heterotrimer composed of three α chains. Formation of these heterotrimers is initiated by C-terminal NC1 domain interactions, accompanied by folding of the collagenous domains into triple helices. There is evidence for at least three types of type IV collagen heterotrimer: α1(IV)2–α2(IV); α3(IV)–α4(IV)–α5(IV); and α5(IV)2–α6(IV). Type IV collagen triple helices form open, nonfibrillar networks that associate with laminin assemblies through interactions mediated by nidogen to form basement membranes. The six type IV collagen genes are arranged in pairs on three chromosomes (Fig. 48-1). The 5′ ends of each gene pair are adjacent to each other, separated by sequences of varying length that contain motifs involved in the regulation of transcriptional activity. There are several distinct type IV collagen networks in basement membranes: a ubiquitous network comprising the α1(IV) and α2(IV) chains; and other networks, restricted in distribution, composed of α3(IV), α4(IV), and α5(IV) chains, or α5(IV) and α6(IV) chains. GBM contains separate α1–α2(IV) and α3–α4–α5(IV) networks, whereas epidermal basement membranes (EBMs) contain separate networks of α1–α2(IV) chains and α5–α6(IV) chains. A network composed of α1, α2, α5, and α6(IV) chains has been described in smooth muscle basement membranes.12 These various networks likely have different physical and functional characteristics and interact differently with other matrix components and adjacent cells. Three forms of AS have been established on a molecular genetic basis: an X-linked form resulting from mutations at the COL4A5 locus, primarily affecting the α5(IV) chain; an autosomal recessive form arising from mutations at the COL4A3 locus or the COL4A4 locus, affecting the α3(IV) and α4(IV) chains, respectively, and an autosomal dominant form from heterozygous mutations in COL4A3 or COL4A4 (Table 48-1). Table 48-1 Molecular genetics of Alport syndrome. X-linked Alport syndrome (XLAS) is the predominant form of the disease, accounting for about 80% of patients. Several hundred COL4A5 mutations have been described, mostly missense mutations, splice-site mutations, and deletions of fewer than 10 base pairs.13,14 A common missense mutation involves replacement of a glycine residue in the collagenous domain of the α5(IV) chain by another amino acid. Such mutations are thought to interfere with the normal folding of the α5(IV) chain into triple helices with other α(IV) chains. Male patients with COL4A5 deletions consistently progress to end-stage renal disease (ESRD) during the second or third decade of life and have deafness.15 This phenotype is associated with most of the missense, nonsense, and splicing mutations of COL4A5 described so far. Several missense mutations of COL4A5 have been associated with late-onset (after the third decade) ESRD and with late development of deafness or normal hearing. The severity of disease in a female heterozygous for a COL4A5 mutation probably depends primarily on the relative activities of the mutant and normal X chromosomes in renal, cochlear, and ocular tissues. Autosomal recessive Alport syndrome (ARAS) arises from mutations affecting both alleles of COL4A3 or COL4A4.15,16 ARAS should be suspected when an individual exhibits the typical clinicopathologic features of the disease but lacks a positive family history, especially when a young female has findings indicative of severe disease, such as deafness, nephrotic syndrome, and impaired renal function, However, sporadic cases of AS may represent de novo mutations at the COL4A5 locus or a germ-line COL4A5 mutation in the proband’s mother. Most patients with ARAS develop ESRD and deafness before age 30 years, regardless of gender. Heterozygous mutations in COL4A3 or COL4A4 typically result in asymptomatic hematuria.16,17 In some families, however, these mutations may also be associated with progressive nephropathy, that is, autosomal dominant Alport syndrome (ADAS).18,19 ADAS patients tend to have a slower course to ESRD than those with XLAS or ARAS.20 The GBMs and tubular basement membranes of males with XLAS usually fail to stain for the α3(IV), α4(IV), and α5(IV) chains but do express the α1(IV) and α2(IV) chains21 (Fig. 48-2). Women with XLAS frequently exhibit mosaicism of GBM expression of the α3(IV), α4(IV), and α5(IV) chains, whereas expression of the α1(IV) and α2(IV) chains is preserved. Most males with XLAS show no EBM expression of α5(IV), whereas female heterozygotes frequently display mosaicism (Fig. 48-3). Lens capsules of some males with XLAS do not express the α3(IV), α4(IV), or α5(IV) chains, whereas expression of these chains appears normal in other patients. In most patients with ARAS, GBM shows no expression of the α3(IV), α4(IV), or α5(IV) chains, but α5(IV) and α6(IV) are expressed in Bowman capsule, distal tubular basement membrane, and EBM20 (Fig. 48-4). Therefore, XLAS and ARAS may be differentiated by immunohistochemical analysis. Basement membrane expression of type IV collagen α chains appears to be normal in patients with ADAS. These observations indicate that a mutation affecting one of the chains involved in the α3–α4–α5(IV) network can prevent GBM expression not only of that chain but also of the other two chains. Most observational and experimental evidence supports the hypothesis that these effects reflect post-translational events. Some mutant chains are unable to participate in the formation of trimers; as a result, the normal chains that are prevented from forming trimers undergo degradation. Other mutations may allow formation of abnormal trimers that are degraded before deposition in basement membranes can occur (Fig. 48-5). Hematuria is the cardinal finding of AS. Affected males have persistent microhematuria. Many also have episodic gross hematuria, precipitated by upper respiratory infections, usually during the first two decades of life. Hematuria has been discovered in the first year of life in affected boys, in whom it is probably present from birth. Boys who are free of hematuria during the first 10 years of life are unlikely to be affected. More than 90% of females with XLAS have persistent or intermittent microhematuria, but about 7% of obligate heterozygotes never manifest hematuria.21 Hematuria appears to be persistent in both males and females with ARAS. About 50% of carriers of COL4A3 or COL4A4 mutations have hematuria.16,17 Proteinuria is usually absent early in life but develops eventually in all males with XLAS and in both males and females with ARAS. Proteinuria increases progressively with age and may result in the nephrotic syndrome. Proteinuria develops eventually in most heterozygous females.22 Hypertension also increases in incidence and severity with age. Similar to proteinuria, hypertension is more likely to occur in affected males than in affected females with XLAS, but there are no gender differences in the hypertension frequency in ARAS. End-stage renal disease develops in all affected males with XLAS, at a rate determined primarily by the nature of the underlying COL4A5 mutation.15 Thus, the rate of progression is fairly constant among affected males within a particular family, but there is significant interkindred variability. Significant intrakindred variability in the rate of progression to ESRD has been reported in some families with missense COL4A5 mutations. Progression to ESRD in females with XLAS was considered an unusual event until a 2003 European study of several hundred XLAS females found that 12% developed ESRD before age 40 (vs. 90% of XLAS males), increasing to 30% by age 60 and 40% by 80.22 The risk for ESRD was significantly increased in heterozygotes with proteinuria. The outcome of XLAS in females is presumed to depend on the relative activities of the normal and mutant X chromosomes, but this has yet to be proved. Gross hematuria in childhood, nephrotic syndrome, and the finding of diffuse GBM thickening by electron microscopy are risk factors for chronic kidney disease (CKD) in affected females.23 Sensorineural deafness and anterior lenticonus are also indicative of an unfavorable outcome in affected women. Both males and females with ARAS appear likely to progress to ESRD during the second or third decade of life. Deafness is frequently but not universally associated with the Alport renal lesion, occurring in about 80% of males and 25% to 30% of females with AS.15,22 In some families with Alport nephropathy and apparently normal hearing, deafness may occur late and may be very slowly progressive. Hearing loss in AS is never congenital and usually becomes apparent by late childhood to early adolescence in boys with XLAS and in both boys and girls with ARAS. Hearing impairment in members of families with AS is always accompanied by evidence of renal involvement. There is no convincing evidence that deaf males lacking renal disease can transmit AS to their offspring. In its early stages, the hearing deficit is detectable only by audiometry, with bilateral reduction in sensitivity to tones in the range 2000 to 8000 Hz. In affected males, the deficit extends progressively to other frequencies, including those of conversational speech. Ocular defects occur in 30% to 40% of XLAS males and in about 15% of XLAS females.15,22 Anterior lenticonus, which is virtually pathognomonic of AS, occurs in about 15% of XLAS males and is almost entirely restricted to AS families with progression to ESRD before age 30 years and deafness.15 Anterior lenticonus is absent at birth, usually appearing during the second to third decade of life, and is bilateral in 75% of patients (Fig. 48-6, A). The spectrum and frequencies of ocular lesions appear to be similar in XLAS and ARAS.24 Another common ocular manifestation of AS is a maculopathy, which consists of whitish or yellowish flecks or granulations in a perimacular distribution and occurs in 15% to 30% of patients (Fig. 48-6, B). The maculopathy does not appear to be associated with any visual abnormalities. Corneal endothelial vesicles (posterior polymorphous dystrophy) have been observed in AS and may indicate defects in Descemet membrane, the basement membrane underlying the corneal endothelium. Recurrent corneal erosion in AS has been attributed to alterations of the corneal EBM. The association of AS with leiomyomatosis of the esophagus and tracheobronchial tree has been reported in about 30 families.14 Affected females typically exhibit genital leiomyomas as well, with clitoral hypertrophy and variable involvement of the labia majora and uterus. Bilateral posterior subcapsular cataracts also occur frequently in affected individuals. Symptoms usually appear in late childhood and include dysphagia, postprandial vomiting, retrosternal or epigastric pain, recurrent bronchitis, dyspnea, cough, and stridor. All patients with AS–diffuse leiomyomatosis complex have been found to have deletions that encompass the 5′ ends of COL4A5 and COL4A6. An autosomal dominant syndrome of hereditary nephritis, deafness, and megathrombocytopenia called Epstein syndrome has been described in a small number of families. Families with Fechtner syndrome exhibit these features as well as leukocyte inclusions (May-Hegglin anomaly). Both Epstein and Fechtner syndrome arise from mutations in nonmuscle myosin heavy-chain IIA.25 Basement membranes of these patients do not exhibit abnormalities in expression of type IV collagen α chains. Therefore, Epstein and Fechtner syndromes are best considered distinct forms of hereditary nephritis rather than variants of AS. Aneurysmal dilation of the thoracic and abdominal aorta and smaller arterial vessels has been described in a small number of males with Alport syndrome.26 There are no pathognomonic lesions by light microscopy or direct immunofluorescence in AS. In affected males, biopsies obtained before 5 years of age typically show no light microscopy changes. Mesangial hypercellularity and matrix expansion are typically observed in older children and adolescents. Glomeruli of affected males eventually show focal segment glomerulosclerosis, and interstitial fibrosis and tubular atrophy are often found in affected boys older than 10. Light microscopy findings in affected females correlate with proteinuria and kidney function; an affected female of any age who has isolated hematuria is likely to have little or no abnormality by light microscopy. Indirect immunofluorescence of type IV collagen α-chain expression in renal or skin basement membranes can be diagnostic (see earlier discussion) and is increasingly available in specialized laboratories around the world. Electron microscopy (EM) frequently reveals diagnostic abnormalities. The cardinal fine structural feature of the kidney that occurs in most patients with AS is the variable thickening, thinning, basket weaving, and lamellation of the GBM (Fig. 48-7). The thick segments measure up to 1200 nm in depth, usually have irregular outer and inner contours, and are found more frequently in males than in females. The lamina densa is transformed into a heterogeneous network of membranous strands, which enclose clear electron-lucent areas that may contain round granules of variable density measuring 20 to 90 nm in diameter. There are variable degrees of epithelial foot process fusion. Not all Alport kindreds demonstrate these characteristic ultrastructural features. Thick, thin, normal, and nonspecifically altered GBMs have all been described. Affected young males, heterozygous females at any age, and, on occasion, affected adult males may have diffusely attenuated GBM measuring as little as 100 nm or less in thickness rather than the pathognomonic lesion. Although diffuse attenuation of GBM has been considered the hallmark of thin basement membrane nephropathy, some patients with this abnormality are members of kindreds with a history of progression to renal failure. Therefore, the significance of an ultrastructural finding of thin GBM must be considered in the context of the family history, basement membrane expression of type IV collagen α chains, and if available, molecular genetic information. Figure 48-8 summarizes the evaluation of patients with hematuria and a positive family history. AS should be included in the initial differential diagnosis of patients with persistent microhematuria after excluding structural abnormalities of the kidneys or urinary tract. The presence on EM of diffuse thickening and multilamellation of the GBM predicts a progressive nephropathy, regardless of family history. However, in a patient with a negative family history, EM cannot differentiate de novo XLAS from ARAS. In some patients, the biopsy findings may be ambiguous, particularly in females and young patients of either gender. Furthermore, families with progressive nephritis and COL4A5 mutations in association with GBM thinning have been described, indicating that the classic Alport GBM lesion is not present in all Alport kindreds.
Alport and Other Familial Glomerular Syndromes
Alport Syndrome
Definition
Etiology and Pathogenesis
Type IV Collagen
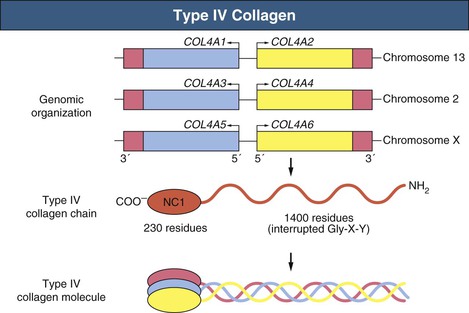
Genetics
Molecular Genetics of Alport Syndrome
Inheritance
Affected Locus
Gene Product
X-linked (XLAS)
COL4A5
α5(IV)
X-linked + leiomyomatosis
COL4A5 + COL4A6
α5(IV) + α6(IV)
Autosomal recessive (ARAS)
COL4A3
α3(IV)
COL4A4
α4(IV)
Autosomal dominant
COL4A3
α3(IV)
COL4A4
α4(IV)
X-Linked Alport Syndrome
Autosomal Recessive Alport Syndrome
Autosomal Dominant Alport Syndrome
Type IV Collagen in Alport Basement Membranes
Clinical Manifestations
Renal Defects
Cochlear Defects
Ocular Defects
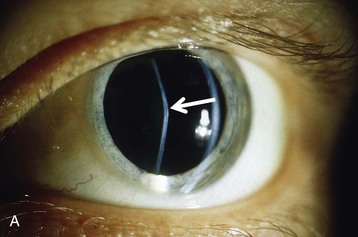
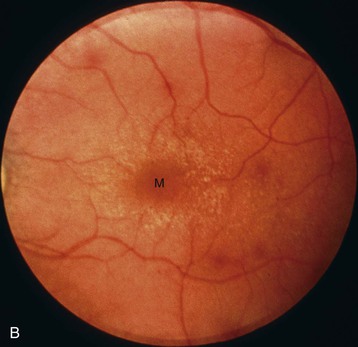
Leiomyomatosis
Hematologic Defects
Arterial Abnormalities
Pathology
Diagnosis and Differential Diagnosis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree