A Clinical Approach to Kidney Disease in Infants, Children, and Adolescents
A Clinical Approach to Kidney Disease in Infants, Children, and Adolescents
Craig B. Langman
Gal Finer
Neziha Celebi
THE EXPRESSION OF “KIDNEY DISEASE” IN CHILDREN
This chapter is intended for medical professionals who are not pediatric nephrologists or pediatricians but who may be called on to evaluate the infant, child, or adolescent patient for the possible presence of kidney disease. Therefore, it is not possible to include an in-depth discussion of all diseases of the specialty and, in particular, the chapter avoids discussions of therapy in most because confirmation of a certain diagnosis of childhood kidney disease would lead the concerned practitioner to consult with a pediatric nephrologist for definitive and ongoing care.
The fundamental difference between pediatric and adult patients is the capacity and, indeed, the expectation of somatic growth and development from infancy through late adolescence, as compared to homeostasis of body mass in the adult. The appearance of kidney disease in childhood interferes with the normalcy of those pediatric processes leading to undisturbed growth (failure to thrive). The pattern of growth from birth through first consultation, plotted formally on available charts of normal patterns of gain in height, height velocity, body mass, and, in children less than 36 months, head circumference, must be a high-ranking task in evaluation of the patient for possible chronic kidney disease (CKD). Absence of changes do not rule out all causes of kidney disease, as discussed throughout the chapter by category of disease, but its presence may help the astute clinician in differential diagnosis.
For ease, and as we teach and practice, children present with a limited, but understandable, series of specific features that alert the clinician to the presence of “kidney disease.” Table 64.1 encompasses all topics discussed in this chapter. Understanding the limitations of the chapter, it is hoped the reader will still find the text useful in everyday consultation and practice.
There are common elements to a history and physical examination of the infant, child, and adolescent when thinking about the presence of kidney disease, and those are summarized in Table 64.2.
Assessment of glomerular filtration rate (GFR) is the single most important test of renal function required in clinical practice. The 24-hour endogenous creatinine clearance is used in adults, but such urine collections can be difficult to obtain in infants and young children, particularly those who are outpatients. The 24-hour urine excretion of creatinine is a measure of creatinine production that is related to muscle mass, and in turn correlates with the cube of height in boys and girls from the age of 6 months to maturity. Because GFR correlates with body surface area (BSA) or the square of height, it follows that GFR corrected for BSA is related to height. Thus,
where V is urine flow rate and Ucr and Pcr are the urine and plasma concentrations of creatinine, respectively. The value for the constant k has been empirically based on measured GFR from iohexol-based GFR studies, and reinterpreted as the creatinine assay itself is now standardized around the world (bedside Chronic Kidney Disease in Children Prospective Cohort Study [CKiD]).
With the newer assays for serum creatinine in infants younger than age 1 year, and in adolescents older than 18 years, Equation 64.5 has not been validated. Due to the difficulty associated with 24-hour urine collection in young children, estimates of GFR from the height and plasma creatinine may be more reliable than the 24-hour endogenous clearance in children and obviously are more convenient. It is important to note that this method is likely unreliable when the normal relation between muscle mass and height is altered, as in malnutrition or muscular dystrophy, or in severe renal failure when tubular secretion of creatinine is increased.
TABLE 64.1 Major Features of the Presentation of Kidney Disease in Childhood
Failure to thrive
Nephrolithiasis
Chronic metabolic acidosis
Rickets
Recurrent volume depletion
Chronic metabolic alkalosis
Polyuria
Kidney Fanconi syndrome
Oliguria
Recurrent urinary infection
Hyponatremia
Hypokalemia
Abdominal masses
Hyperkalemia
Dysuria, frequency
Edema
Disorders of sexual differentiation
Hematuria
Proteinuria
Hypercalcemia
Hypertension
Hypocalcemia
Hypophosphatemia
Hypomagnesemia
Hyperphosphatemia
Thrombotic microangiopathy
Hypocomplementemia
Plasma creatinine concentration averages 0.88 mg per dL at birth, when the level is largely determined by the mother’s plasma creatinine concentration. It falls to a nadir of 0.32 mg per dL at 2 years as GFR increases and then rises with the increase in muscle mass. From age 2 weeks to 5 years, a normal blood creatinine level is between 0.11 and 0.35 mg per dL. From 5 to 10 years, a normal blood creatinine level is between 0.28 and 0.55 mg per dL. Normal serum creatinine may be as high as 0.84 to 0.93 mg per dL in older teenagers or adult men. However, a serum creatinine concentration greater than 1.10 to 1.20 mg per dL should raise concern for underlying renal disease. Clinical paradigms for measured GFR (mGFR) using iohexol are evolving quickly in the pediatric nephrology clinics.
The addition of other agents such as cystatin C into the estimated GFR (eGFR) equation may improve precision, but not offer substantial important information when the mGFR is above 75 mL/min/1.73 m2. Adolescents of adult size may benefit from the MDRD equation used in adults for determination of eGFR.
The expression of monogenic mutations is not infrequent in a variety of chronic kidney diseases in pediatrics, and encompasses the entire clinical spectrum of such disorders, from congenital anomalies of the kidney and urinary tract (CAKUT), glomerular and tubular disorders, through specific transport defects of single, selected ions, minerals, or other substances. As a result of the sequencing of the human genome, and the use of advanced techniques including high throughput technologies, rapid discoveries are made daily. As a result of the rapidity of mutational gene discoveries linked to pediatric kidney disease expression in patients, a comprehensive listing is immediately out of date. However, up-to-date knowledge of such findings can be learned from Online Mendelian Inheritance in Man (OMIM; http://www.omim.org/). Currently known mutations for monogenic disorders and CAKUT are shown in Tables 64.3 and 64.4, respectively. Throughout this chapter, diseases referred to may be found in this listing with a hyperlink about them.
Embryogenesis of the kidney and urinary tract has been advanced at the molecular level considerably in the recent past, and explains, in part, many of the well-recognized malformation complexes seen in patients ranging from infancy (e.g., aplasia, hypoplasia, dysplasia, cystic diseases) through later adolescence (e.g., autosomal-dominant polycystic kidney disease, autosomal-recessive nephrolithiasis with kidney failure/Dents disease). Alternatively, sporadic urologic malformations, the third most common birth defect overall in live-born neonates (e.g., posterior urethral valves, hydronephrosis), may not have easy to decipher molecular pathophysiology, but must be remembered not only in infancy, but during the entire spectrum of pediatrics, as their consequences may only be revealed in the older child or adolescent with the appearance of progressive CKD.
As in adult CKD, polygenic factors play a role in the expression of other disorders, or in their severity of expression. Little information that is different for pediatric CKD when compared to adult disease is available now, and the reader is referred to other chapters in this volume for a discussion of the topic.
HEMATURIA
Hematuria is one of the most common urinary symptoms in children, and may represent a benign condition such as hematuria resulting from strenuous exercise or from a life threatening illness such as Wilms tumor. Hematuria originating from glomerulonephritis (GN) is often signified by coco-cola- or tea-colored urine, red blood cell casts, and/or dysmorphic red blood cells (RBCs), whereas bleeding distal to the glomerulus, such as from urologic issues or infections, are more likely to be associated with red urine and end of void hematuria.
History
Abdominal, flank, or groin pain is suggestive of nephrolithiasis. Dysuria, foul smelling urine, urgency, and increased voiding frequency with or without fever may denote a urinary tract infection (UTI) that is usually caused by gram-negative bacteria or adenovirus in children outside of infancy or, alternatively, may represent the presence of hypercalciuria. Abdominal trauma may induce hematuria especially in abnormally shaped kidneys of polycystic or hydronephrotic nature. In postinfectious GN there is a window of at least a week between the occurrence of the febrile disease, most commonly streptococcal pharyngitis or impetigo, to the onset of tea-colored urine, whereas in immunoglobulin A (IgA) nephropathy, another common cause of acute glomerulonephritis in children, a febrile prodrome precedes renal symptoms by only 2 to 3 days. The review of systems is imperative in a child with hematuria as the differential diagnosis includes systemic lupus erythematosus (SLE) and other vasculitides. Alport syndrome, sickle cell disease, and benign familial hematuria are examples of hereditary conditions associated with microscopic hematuria. It is very unlikely for coagulopathic states to manifest as hematuria alone, although a history of bleeding disorder should be sought.
TABLE 64.2 Common Elements of the Pediatric History and Physical Examination
History
Physical Examination
Birth History: complete maternal obstetric history; birth weight, length, and head circumference; gestational age; placental abnormalities; neonatal history of illness and medications; malformations outside the kidney
Growth percentiles for length, body mass (weight), head circumference (through 2 years of age; http://www.cdc.gov/growthcharts/)
Feeding and Dietary History: source of protein intake (breast, formula); food allergies; food avoidances; urination pattern (frequency, stream appearance); defecation pattern
Blood pressure with appropriate size cuff, and if concern over hypertension, four extremity blood pressure at first consultation; plot normative values for measured arm blood pressure for boys and girls, respectively:
Family History: chronic kidney diseases, kidney rransplantation; hypertension; cystic kidney diseases; kidney stones; infant deaths; fractures in young adults; early onset osteoporosis; “unusual diseases”; early onset myocardial infarction, stroke
Complete Physical Examination, with special emphasis depending on the reason for consultation to: skin lesions, rashes, or purpura; peripheral edema; retinal examination for pigmentary changes, anterior chamber for crystal deposits, abnormalities of the eye; alterations of ears (location on the skull relative to the palpebral fissures; size and/or malformations); vascular bruits including abdominal aorta, renal artery locations; abdominal masses; organomegaly; sexual development, feminization, or masculinization of the opposite gender, Tanner sexual maturity rating; presence of arthritis or arthralgias; muscle tone; numbers of fingers and toes; scoliosis; rachitic changes of the extremities, chest and/or skull
Complete Review of Ten Systems
Social History: developmental milestone achievements; school performance
A listing of relevant milestones by age can be accessed here: http://www.cdc.gov/ncbddd/actearly/milestones/
Immunization History
Schedules and recommendations by age can be accessed here:
Periorbital and facial edema are suggestive of GN. The joints and skin should be examined for involvement in vasculitis. Intravascular volume contraction, especially in infancy, can predispose to renal vein thrombosis that may present with gross or microscopic hematuria.
Reduced number of UB branches and nephrons that are fully formed, small kidney size
Pax2, Sall1 Six2, BMP4 HNF1β UMOD
Renal dysplasia
Aberrant interactions among the UB, MM, or stroma
Reduced number of UB branches and nephrons. Presence of undiffirentiated stromal and mesenchymal cells, cysts, or cartilage. Frequently associated with kidney hypoplasia
Pax2 HNF1β UMOD Nphp1 BMP4, Six2 XPNPEP3
Polycystic kidneys
Aberrant tubular and collecting duct patterning
Cysts in tubules and collecting ducts Normally formed glomeruli
Pkd1, Pkd2 HNF1β HPHP1
Multicystic dysplastic kidneys
Aberrant interactions among the UB, MM, or stroma
Absence of glomeruli and tubules Presence of large cysts Aberrant patterning Poorly formed atretic ureters Small remnant kidney (if organ involutes)
HNF1β UPIIIA
Medullary cystic kidney disease 2
Aberrant tubular and collecting-duct patterning
Tubular atrophy, interstitial fibrosis, cysts in distal tubules and medullary collecting ducts
UMOD
Duplex ureters
Supernumerary UB budding from the ND
Duplex ureters and kidneys or duplex ureters and collecting systems
May be associated with VUR or obstruction if UB budding is ectopic
Robo2
FoxC1
FoxC2
BMP4
Horseshoe kidney
Defects in renal capsule
Kidneys are fused at inferior lobes and located lower than usual
HNF1β
UB, ureteric bud; MM, metanephric mesenchyme; ND, nephric duct.
Laboratory
In evaluation of hematuria, the urine should be examined both by dipstick and under a microscope. A positive urine dipstick for blood in the absence of RBC under the microscope suggests other causes of pigmenturia, such as hemoglobinuria or myoglobinuria. The presence of proteinuria (the dipstick is qualitative) is consistent with GN. Hematuria with low complement levels should raise the diagnosis of postinfectious GN, atypical hemolytic uremic syndrome, membranoproliferative GN, and GN associated with ventricular-peritoneal shunt infection and endocarditis. Increased calcium to creatinine ratio (>0.4) may signify the presence of hypercalciuria, a common condition leading to microscopic hematuria in childhood, and prompt an evaluation discussed in that section of this chapter.
Imaging
A kidney ultrasound is mandatory in the evaluation of hematuria, and further imaging should be ordered as required by the working diagnosis. As a general rule, intravenous pyelography has been replaced by other imaging modalities. Care should be taken in ordering imaging beyond the kidney ultrasound because all the side effects seen in adults with kidney disease (e.g., contrast nephropathy; nephrogenic systemic fibrosis) may occur in the pediatric population as well. Lifetime exposure to radiation factors into the decision as well for the pediatric patient undergoing such imaging. Rare causes of hematuria in children include abdominal tumors (neuroblastoma and rhabdomyosarcoma), or arteriovenous malformations of the kidney vasculature. Red urine with negative dipstick can be encountered in neonates as a result of urate crystals excretion (“brick dust” urine), and at all ages as the result of medications (e.g., rifampin, phenazopyridine), foods such as beets, or aniline dyes.
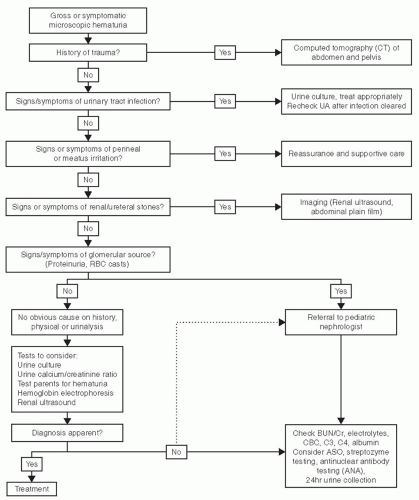
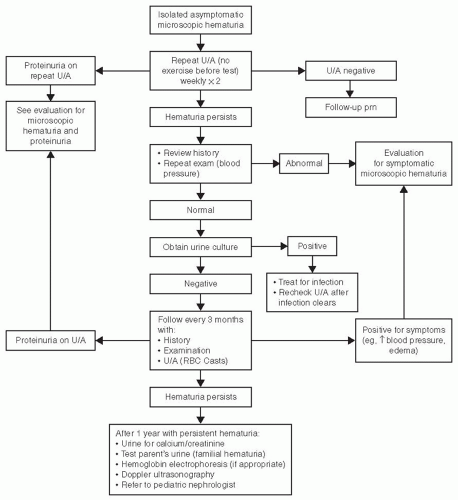
Figures 64.1 and 64.2 give a reasonable approach to the evaluation of microscopic and gross hematuria, respectively.
PROTEINURIA
Quantitative assessment of proteinuria in children is affected by the unreliability of extended urine collections. However, its main purpose is to detect damage to the glomerular filter leading to increased permeability, and for this purpose the most sensitive parameter is the sieving coefficient (the relative concentration in glomerular filtrate and plasma water) of a molecule of a size that normally is just restrained by the glomerular filter. The urinary protein to urinary creatinine ratio provides a suitable approximation for use in clinical practice; being a ratio, it is independent of urine flow rate and can be estimated from a random urine sample. A value of less than 0.2 is considered normal and greater than 2.0 is considered nephrotic range proteinuria in children. The use of this ratio is preferred for children rather than the commonly performed 24-hour urine excretion for which more than 200 mg or 5 mg per kg per day usually is regarded as abnormal in children.
Normal urine contains a minimal amount of protein and is physiologic. Increased urine protein can be an isolated finding in a benign condition such as orthostatic proteinuria, or associated with significant kidney disease as nephrotic syndrome or CKD.
Definition
The urine dipstick is a sensitive screening tool for albuminuria and the presence of > 1 + indicates a further diagnostic workup is prudent. A 24-hour urine collection is considered the gold standard method to quantify protein excretion, but is very difficult to achieve in children. When collected, values above indexed to body surface area (m2). Values above 4 mg/m2/h are pathologic. Values above 40 mg/m2/h signify nephrotic range proteinuria. Because of the difficulty in performing the 24-hour urinary collection for protein, a random urine sample for protein (mg/dL) and creatinine (mg/dL) ratio (unitless), preferably on the first morning void, is commonly obtained. A ratio above 0.2, beyond the first year of life, is abnormal and warrants evaluation. Neonatal urine contains higher levels of albumin and lower molecular weight proteins, so an albumin/creatinine ratio is more commonly obtained.
History and Physical Exam
Febrile illness, strenuous exercise, and severe dehydration are commonly associated with transient proteinuria that has no long-term renal consequences. The presence of facial, periorbital, pedal, and/or scrotal edema are suggestive of low serum albumin and nephrotic syndrome. A thorough review of systems and a family history of kidney diseases are always important in the face of proteinuria.
Laboratory
The nature of the protein found in the patient’s urine may imply the location of the renal lesion; glomerular diseases usually result in albuminuria whereas tubular pathology often leads to low molecular weight proteinuria such as beta-2-microglobulin. The finding of tubular proteinuria may be seen in the setting of generalized proximal tubular disorders that can also lead to aminoaciduria, phosphaturia, glucosuria, and/or bicarbonaturia (Fanconi syndrome). Low-molecular-weight proteinuria has also been linked to renal interstitial damage related to reflux nephropathy, obstructive nephropathy, or acute and chronic pyelonephritis. In orthostatic proteinuria the first morning urine protein to creatinine ratio is normal. In this condition a timed 24-hour urine collection will result in elevated protein excretion when the subject is upright with normal urine protein in recumbency. Nephrotic range proteinuria in the setting of peripheral edema, hypoalbuminemia, and hypercholesterolemia are diagnostic of nephrotic syndrome (see section on nephrotic syndrome for further evaluation). Many of the conditions leading to CKD are associated with proteinuria. In CKD, proteinuria itself is thought to be a perpetuating factor, leading to further damage by various mechanisms including induction of apoptosis, cell atrophy, and epithelial to mesenchymal transformation. Isolated proteinuria in repeated urine samples over time is always an index of renal abnormality and should be further investigated. In addition to urinalysis, kidney function and serum electrolytes are required measurements in almost all cases of proteinuria. Persistent proteinuria of unclear etiology warrants the consideration of a kidney biopsy.
FIGURE 64.1 Approach to evaluation of gross hematuria.
Table 64.5 lists a classification and some of the many causes of proteinuria in children.
ACUTE GLOMERULONEPHRITIS
The sudden onset of gross hematuria, proteinuria, azotemia, edema, and hypertension is a classic description of acute glomerulonephritis (AGN) in children and adolescents (Table 64.6). An important nodal point in the differential diagnosis is the level of serum complement factors, C3 and C4, which when depressed, suggest postinfectious (poststreptococcal disease most commonly) AGN, membranoproliferative GN, subacute bacterial endocarditis, shunt nephritis (plastic devices such as drains for cerebrospinal fluid placed into the vascular space), or, rarely, quartan malarial GN. Normocomplementemic postinfectious AGN, although occurring in up to 10% of those with biopsy-proven postinfectious AGN, suggests other etiologies that include IgA GN, Henoch-Schönlein purpura (when accompanied by a purpuric, lower extremity rash), vasculitides that include collagen-vascular diseases such as SLE, granulomatosis with polyangiitis (Wegeners), Goodpasture syndrome, or pauci-immune vasculitis.
FIGURE 64.2 Approach to the evaluation of microscopic hematuria.
Postinfectious AGN when associated with other components of thrombotic microangiopathies, including anemia, thrombocytopenia, and other wide-ranging organ dysfunctions, may connote diseases such as thrombotic thrombocytopenic purpura (TTP), shiga-toxin associated enterocolitis hemolytic uremic syndrome (STEC-HUS), or atypical HUS, a disease of complement regulatory protein mutational abnormalities, or in combination with autoantibodies to some of those proteins, in which the alternative complement pathway is constitutively activated and unregulated, leading to overactivity and resultant disease.
As in adults, a presentation with rapidly advancing kidney failure may be associated with a crescentic presentation on kidney biopsy that defines rapidly progressive glomerulonephritis (RPGN), most commonly associated in children with postinfectious AGN from postinfectious GN or one of the vasculitides noted.
TABLE 64.5 Classification and Causes of Proteinuria in Children
Transient Proteinuria, associated with fever, vigorous exercise, volume depletion, or prolonged seizures
Orthostatic Proteinuria, in which the first morning urine is devoid of pathologic levels of protein but subsequent urines throughout the day reveal excess protein levels
Glomerular Proteinuria, arising from any form of infantile or childhood nephrotic syndrome, or as part of a glomerulonephritis of any etiology
Tubular Proteinuria, associated with primary or acquired diseases of the proximal tubule, obstructive uropathy, or tubulointerstitial nephritis (including pyelonephritis)
Diagnostic evaluation of postinfectious AGN in children and adolescents should include evaluation for recent streptococcal infection of the throat (anti-streptolysin O titer), skin (anti-DNaseB titer), collagen-vascular disease, ANCA titers, complement levels, and a search for thrombotic microangiopathy (complete blood count [CBC], peripheral blood smear for schistocytes, reticulocyte count, platelet count, lactate dehydrogenase [LDH], haptoglobin level), and imaging of the kidneys by ultrasound to evaluate size and echo texture.
A percutaneous kidney biopsy in children and adolescents is a safe procedure performed by those with biopsy skills and the ability to provide proper anesthetic management. The biopsy is processed routinely for light, immunologic, and electron microscopic examinations and should be interpreted in collaboration with a pathologist skilled in pediatric kidney diseases.
As a general rule, the diagnosis of hypocomplementemic, postinfectious AGN in the child or adolescent does not mandate a kidney biopsy, as the hypocomplementemia resolves in 6 weeks in the overwhelming majority of cases, and the disease carries an excellent prognosis overall. Other causes of postinfectious AGN may indeed warrant a kidney biopsy early in its course for diagnostic, prognostic, and therapeutic decisions.
From young childhood (older than three years) through late adolescence, the sudden appearance of gross hematuria with microscopic RBC casts, proteinuria, edema, and hypertension approximately 1 week to 1 month following a streptococcal pharyngitis or skin-based infection have defined the occurrence of postinfectious (poststreptococcal) postinfectious AGN. Documentation of a positive throat or skin culture for group A β-hemolytic streptococcus, or serologic evidence of elevated antistreptolysin O or DNaseB titers is commonly found and strengthens the diagnosis of postinfectious AGN. In developing areas of the world, many additional causative agents have been identified including other bacteria, viruses, fungi, and parasitic infectious agents.
TABLE 64.6 Common Causes of Acute Glomerulonephritis in Children and Adolescents
Associated with Hypocomplementemia
Associated with Normal Levels of Complement
Postinfectious AGN
GPA (granulomatosis with polyangiitis); MPA (microscopic polyangiitis); PAN (polyarteritis nodosa)
The differential diagnosis of postinfectious AGN includes any kidney disease in which there is a nephritic picture, including IgA nephropathy, Henoch-Schönlein purpura, Goodpasture syndrome, systemic vasculitis related to ANCA, SLE, forms of hemolytic uremic syndrome, membranoproliferative glomerulonephritis, or an acute presentation of other chronic glomerular diseases.
Group A β-hemolytic streptococcus of the M-type is most commonly associated with the occurrence of postinfectious AGN, and type 12 is commonly isolated in cases of postinfectious AGN related to pharyngitis, and type 49 is commonly isolated in postinfectious AGN after skin-based infections. These are termed nephritogenic, although other types may produce postinfectious AGN too.
Postinfectious AGN is a prototypic hypocomplementemic disease in which there appears to be passive antigenantibody complex deposition within the glomerulus in a characteristic subepithelial location, although many variants have been described that may mimic either membranoproliferative glomerulonephritis type I or, C3-glomerulopathy on kidney biopsy. The exact mechanisms whereby the circulating immune complexes deposit within the glomerulus remain uncertain, but may involve the alternative complement pathway (low C3, normal C4, and no C1q deposition in the kidney), rather than the classical complement pathway.
The hypocomplementemia is of short duration, with over 95% of patients recovering normal levels of complement C3 6 weeks after presentation. Postinfectious AGN may present with an RPGN clinical picture as well, with intense crescent formation found in the kidney biopsy. The depth of C3 depression does not correlate with disease severity or course. Up to 10% of patients with postinfectious AGN have normal C3 levels, depending on the time at which it is measured.
A kidney biopsy is rarely performed for postinfectious AGN related to group A β-hemolytic streptococcus, but is reserved for RPGN, uncertain assignment of postinfectious AGN as to etiology, or cases in which systemic manifestations suggest another cause of the postinfectious AGN.
More subtle cases of postinfectious AGN may be seen when the streptococcal disease is of a more epidemic nature, and in which gross hematuria is often absent and azotemia is minimal. We recommend a screening urinalysis of family members of index cases of postinfectious AGN, looking for hematuria and proteinuria, whether the case is epidemic or isolated.
No specific therapy has been shown to change the course of the typical child with postinfectious AGN, but we recommend looking for group A β-hemolytic streptococcus by culture, since treatment is warranted to prevent rheumatic fever if infection is present. The child with postinfectious AGN must be evaluated and managed for the potential complications of AGN that include malignant hypertension and its systemic sequelae, oligoanuria with hyperkalemia, dilutional hyponatremia, and acidosis. Rarely such management includes the use of iterative dialysis, unless a rapidly progressive picture is present and severe.
The prognosis of the overwhelming majority of isolated cases of postinfectious AGN that are related to an antecedent group A β-hemolytic streptococcal infection and without a picture of RPGN is uniformly excellent, with full recovery of normal kidney function and no long-term sequelae in over 95%. In developing countries, and in cases where prolonged dialysis is needed for RPGN, the prognosis is less well established, and continuation into ESRD or death may occur.
The triadic finding of a predominant lower extremity and buttocks purpuric rash, abdominal pain, and fever in children and adolescents is prototypic for Henoch-Schönlein purpura (HSP). The disease is classified as a small vessel vasculitis of unknown etiology and, in addition to the organs involved above, may involve the kidney, the lower urinary tract, the joints, and, less commonly, the central nervous system or other organs. The disease is self-limited, may be recurrent, and the long-term morbidity is related to the degree, if any, of kidney involvement.
There is a literature that HSP is related to an antecedent infection, but no specific ones are implicated. Prior drug exposures have been noted rarely, and do not explain the overwhelming majority of such cases. Despite the presence of IgA deposition in the kidney, when kidney involvement is detected, circulating IgA levels are normal. Some evidence for circulating immune-complexes directed against altered moieties of IgA with subsequent mesangial deposition appears.
Kidney involvement is variable, and case series for frequency of involvement may reflect selection and referral biases. Even when initially absent as demonstrated by a normal urinalysis and serum creatinine initially, the kidney may become involved with a subsequent recurrence of the disease rash, but the exact frequency of such involvement subsequently ranges from infrequent to frequent for the biases cited. A good rule of thumb is that the absence of kidney involvement 2 months after clinical presentation of HSP (and without HSP recurrence) is generally associated with long-term sparing of kidney disease at all. Therefore, monitoring the child with HSP in those first 8 weeks after presentation with HSP, including a urinalysis, kidney function, and blood pressure measurement, seems warranted.
Kidney involvement, when present, ranges from lowgrade proteinuria and hematuria, to AGN, or a nephrotic syndrome with heavy hematuria. A kidney biopsy is warranted for the latter three presentations for diagnosis, where a large range of findings may occur, from very little light microscopic abnormalities through focal and segmental proliferative GN and, at the worst, a crescentic GN. IgA is found commonly in association with IgG, C3, and fibrin in a granular, mesangial pattern that is indistinguishable from primary IgA nephropathy. HSP requires, therefore, the presence of the extrarenal system involvement as noted previously.
There is no specific treatment for HSP in general, and even the use of corticosteroids for the extrarenal manifestations remains unproven. In general, with general supportive measures the extrarenal disease improves and has no long-term sequelae in most.
In literature case series, the absence of kidney involvement 6 months after presentation is associated with a uniformly good outcome for normal kidney function. If severe manifestations of kidney involvement in HSP should occur the prognosis for good outcome diminishes, and should be followed for the appearance and/or management of CKD.
Infants, children, and adolescents may present with a thrombotic microangiopathy (TMA) involving the kidney. TMA-based syndromes (Table 64.7) are microvascular occlusive disorders that result from aggregation of platelets, thrombocytopenia, and mechanical injury to erythrocytes, ultimately leading to organ dysfunction. A more specific definition of TMA is the activation of the endothelium due to various insults followed by a cascade of pathologic responses, including among others, platelet and/or complement activation of the terminal (C5b-9) complex, microthrombi formation, thrombocytopenia, and microangiopathic hemolytic anemia. Atypical hemolytic uremic syndrome (HUS) occurs both in the pediatric and adult populations; severe kidney impairment is a prominent but not an essential feature of the disease.
The most common cause of TMA and HUS is that associated with shiga-toxin producing infections (now termed STEC-HUS), including Escherichia coli serotype 0157:H7 and Shigella dysenteriae serotype 1, accounting for more than 90% of HUS cases. Recently, an outbreak in adults was linked to a unique serotype of E. coli, O4:H4. New evidence points to complement activation in STEC-HUS. The term atypical HUS has been used to describe cases of TMA not caused by shiga-toxin associated microorganisms and bacterial infections in general, and in which thrombotic thrombocytopenic purpura (TTP) has been excluded in the differential diagnosis by demonstrating levels of ADAMTS13 activity above 5% to 10%.
TABLE 64.7 TMA-Based Diseases
▪ Thrombotic thrombocytopenic purpura
▪ Congenital ADAMTS13 deficiency
▪ Antibody-mediated ADAMTS12 deficiency
▪ Hemolytic uremic syndrome
▪ STEC-HUS
▪ Pneumococcal HUS
▪ Atypical HUS (atypical HUS)
▪ TMA associated with
▪ Medications
▪ Hypertension
▪ Pregnancy/HELLP syndrome
▪ Solid organ transplantation
▪ Stem cell transplantation
▪ Malignant solid tumors
▪ HIV
▪ Vasculitis, such as systemic lupus erythematosus
Atypical hemolytic uremic syndrome is a rare, life-threatening, chronic, genetic disease of uncontrolled alternative pathway complement activation. The understanding of the pathophysiology and genetics of this disease has expanded over recent decades and promising new developments in the management of atypical HUS have emerged.
In 50% to 60% of cases of atypical HUS, a genetic mutation in complement regulatory proteins and/or autoantibodies against these proteins has been found as an explanation for constitutive complement activation. Mutations have been described in complement factor H (CFH), complement factor I (CFI), Membrane Cofactor Protein (MCP), complement factor B (CFB), myosin binding protein C3 gene (C3), and thrombomodulin. In addition, autoantibodies to CFH can cause atypical HUS and are commonly associated with deletions of complement factor H-related proteins CFHR1 and CFHR3. Mutations in either CFH, CFI, MCP, thrombomodulin, and/or CFHR1/3 with autoantibodies to CFH are associated with loss of regulatory control of the alternative pathway of the complement cascade. Mutations in CFB and C3 are gain of function mutations leading to complement over-activation. A very rare cause of atypical HUS is genetic deficiency of cobalamimase activity.
However, this leaves another 40% to 50% of patients in whom a mutation or autoantibody cannot be demonstrated but for whom atypical HUS is the diagnosis. Thus, the ultimate diagnosis of atypical HUS does not require a formal demonstration of its underlying genetic cause. Less than 20% of atypical HUS cases are familial, with both autosomal-dominant and autosomal-recessive inheritance reported. Autosomal-recessive cases tend to present in childhood whereas autosomal-dominant cases more typically present in adulthood—prognosis is poor regardless of inheritance. Identification of a genetic mutation, although not required for an individual’s diagnosis or management of atypical HUS, may be helpful for identifying and monitoring disease carriers and for providing genetic counseling.
Loss of function mutations in CFH are most common and have the worst prognosis based on registry data, with 60% to 70% of patients progressing to end-stage renal disease (ESRD) or death within a year of disease onset. The ultimate kidney and patient-based prognoses for patients with CFI mutations appears slightly better, followed by patients with MCP mutations, of whom 20% require renal replacement therapy. Patients without demonstrated mutations have similar dire outcomes, however, raising the idea that the presence or absence of a given mutation may have limited prognostic value, except for the MCP mutation that may not recur after kidney transplantation.
Until recently, there have been no specific therapies for atypical HUS. Therapeutic plasma exchange or plasma infusion has generally been the initial approach to disease management, although there are no randomized controlled trials of plasma therapy in atypical HUS to establish its effectiveness. Plasma exchange may only be beneficial for atypical HUS in the short term, because long-term kidney outcomes are uniformly poor with a varying short-term response in hematologic parameters. Plasma exchange would not be expected to be effective for patients with mutations in MCP, a transmembrane protein. Patients who do respond to plasma exchange frequently become plasma dependent, requiring long-term therapy to maintain remission.
Kidney transplantation can be successful for patients with MCP mutations. MCP is cell membrane-bound and highly expressed in the kidney; kidney transplant, then, would be expected to halt the disease process. Other mutations or unknown ones have led to high relapse rates of atypical HUS in the transplanted kidney. CFH and CFI mutations have been studied more extensively. These circulating proteins are primarily synthesized in the liver. Not unexpectedly, atypical HUS recurs in 80% of patients with CFH mutations and 90% of patients with CFI mutations after an isolated kidney transplant. Living-donor kidney transplantation is contraindicated in patients with atypical HUS due to mutations in CFH, CFI, C3, and CFB without other therapies concomitantly.
Combined liver-kidney transplantation has been attempted for patients with CFH and CFI mutations to address the abnormal protein synthesis in the liver and its downstream effect on the kidney. Simultaneous liver-kidney transplantation with prophylactic use of plasma therapy has been successful in patients with CFH mutations. However, liver-kidney transplantation is associated with a higher mortality rate than kidney transplantation alone. In the absence of a noted mutation, comprising a sizable fraction of patients with atypical HUS, liver-kidney transplantation should be avoided.
A pathophysiologic-based treatment in atypical HUS is available now with eculizumab, through inhibiting the formation of the common terminal complement complex (C5b-9). Its recent approval for atypical HUS in adults and in children represents its first approved use for pediatric patients. Due to the impaired capacity for opsonization and clearance of encapsulated organisms, meningococcal disease is a risk with the use of eculizumab and has been reported among patients given eculizumab for paroxysmal nocturnal hemoglobinuria (PNH). Patients must receive the meningococcal vaccine prior to treatment initiation.
The classic triad of findings in nephrotic syndrome in children, adolescents, and adults—hypoalbuminemia, high-degree proteinuria, and edema—are found in infants with nephrotic syndrome too. Congenital nephrotic syndrome has been used to describe the disease in utero through the first 3 months of life, whereas infantile nephrotic syndrome has referred to the development of the disease in months 4 through 12. We find this an arbitrary division, as there is great overlap in the causes between the two groupings, and prefer nephrotic syndrome in the first year of life (NSFL).
The differential diagnosis of NSFL includes disease secondary to congenital maternal infections (e.g., syphilis, cytomegalovirus) or from genetic causes. The majority of the genetic causes involve mutations in kidney morphogens such as WT-1, or mutations in genes related to the integrity of the tri-partite glomerular filtration barrier (the podocyte foot processes, the fenestrated glomerular endothelium, and the glomerular basement membrane). After exclusion of infection-related NSFL, NSFL is now considered a monogenic disease, with one of four genetic mutations (nephrin, podocin, WT1, LAMB2) accounting for over 66% of the cases in a recent Western European series. The disease is not responsive to corticosteroids or immunosuppressive medications as a general principle. Prompt referral to a center skilled in management of infantile nephrosis is recommended.
NSFL may be seen in syndromic diseases of newborns as well, and the clinician should evaluate the patient for extrakidney malformations including disorders of sexual differentiation (see below). For a comprehensive listing, the reader can access: http://www.ncbi.nlm.nih.gov/omim? term=nephrotic%20syndrome%20in%20infancy.
Primary nephrotic syndrome in children from 15 months through 8 to 10 years of age includes minimal change nephrotic syndrome (MCNS), focal segmental glomerulosclerosis (FSGS), membranoproliferative glomerulonephritis (MPGN; types I, II, III), and membranous nephropathy (MN). The overwhelming majority of children will have MCNS when the patient has normal kidney filtering function, is nonazotemic, normotensive, and with normocomplementemic. To date, the initial response to a course of oral corticosteroids, 2 mg/kg/day or 60 mg/m2 BSA/day, defines the disease as steroid-responsive or resistant. Initial corticosteroid resistance mandates a biopsy for histologic diagnosis.
Only gold members can continue reading. Log In or Register to continue
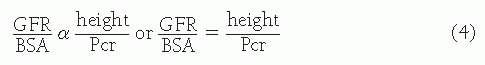
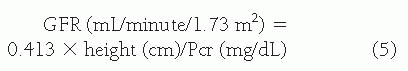
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis



