Infectious conditions
Focal pyelonephritis
Renal abscess
Xanthogranulomatous pyelonephritis
Malakoplakia
Aspergillosis
Echinococcus
Upper tract fungal infection
Cystic lesions
Acquired cystic kidney disease
Polycystic kidney disease
Miscellaneous lesions
Renal-adrenal fusion
Ectopic adrenal tissue
Splenorenal fusion
Extramedullary hematopoiesis
Inflammatory and Infectious Lesions
Focal (Localized) Pyelonephritis
Focal pyelonephritis refers to the infection of a single lobe of the kidney which, in contrast to more diffuse infection, can mimic a renal mass on imaging, especially in the setting of soft tissue extension of the inflammatory process (Fig. 3.1) [1]. Contrast-enhanced computed tomography (CT) is the gold standard for diagnosis of focal pyelonephritis when suspected. On CT, the lesion will appear as a hypoattenuating, poorly circumscribed, or wedge-shaped lesion. Ultrasound may provide a useful ancillary imaging modality and may demonstrate generalized renal enlargement with or without an ill-defined focal lesion [2, 3].
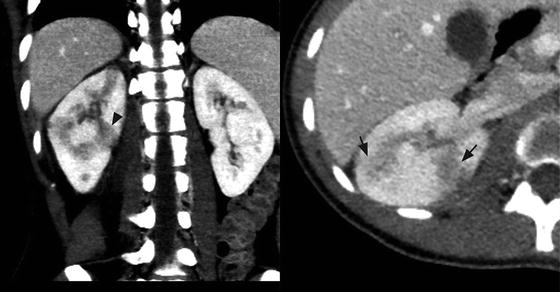
Fig. 3.1
Focal pyelonephritis. Contrast-enhanced CT of the right kidney shows geographic areas of poor cortical enhancement (black arrows) that has a wedge-shaped configuration (black arrow head)
Focal pyelonephritis typically occurs in children and immunocompromised patients, and the diagnosis should be suspected in patients with fever, chills, or flank pain (Table 3.2). Other findings such as nausea, vomiting, and lower urinary tract symptoms (frequency, urgency, or dysuria) can occur. Physical exam may reveal abdominal pain and/or costovertebral angle tenderness in association with fever [4]. Leukocytosis and an elevated C-reactive protein are frequently demonstrated on laboratory evaluation and urine, and blood cultures are commonly positive. The vast majority of responsible pathogens are gram-negative bacteria, and E. coli is the most commonly isolated organism [5]. As pyuria may be absent in 50–70 % of patients, a normal urinalysis does not exclude the diagnosis in the appropriate clinical setting [5, 6]. Biopsy is not typically required in the setting of classic symptoms; however, in instances in which a biopsy is performed, a mixed acute and chronic inflammatory infiltrate may be seen. However, giant cells are not typical.
Table 3.2
Symptoms and signs of infectious process
Symptoms |
Fevers |
Chills |
Flank or abdominal pain |
Nausea, vomiting |
Lower urinary tract symptoms (dysuria, frequency) |
Malaise |
Fatigue |
Anorexia |
Signs |
Fever |
Tachycardia |
Diaphoresis |
Abdominal tenderness |
Costovertebral angle tenderness |
Weight loss |
Palpable mass |
Fistulous tract |
Skin infection |
Diagnosis and aggressive treatment is critical in these cases, as the outpatient therapy typically used for acute uncomplicated pyelonephritis is rarely effective in these cases. Furthermore, hospitalizations may be prolonged due to persistent fever. The recommended duration of intravenous and oral antimicrobial treatment is at least 3 weeks [7].
Renal Abscess
Renal abscess refers to a discrete collection of purulent material originating from the renal parenchyma. This is in contrast to focal pyelonephritis, which but does not present with a collection of fluid and/or debris within the renal parenchyma. While this condition can present at any age, most published literature examines the pediatric experience. The majority of cases occur secondary to ascending infection of the urinary tract, although spread to the kidney may also occur via a hematogenous route. The clinical presentation is similar to focal pyelonephritis with fever, chills, and flank pain (Table 3.2); laboratory evidence of infection, including leukocytosis and positive blood cultures, is evident [8, 9]. Common etiologic pathogens include E. coli, P. mirabilis, and S. aureus, although other less common bacteria have also been documented [1, 10].
On imaging, these lesions have variable characteristics dependent on the phase of infection (early vs. late) and the amount of solid debris within the abscess (Fig. 3.2). Ultrasound and CT are the most commonly utilized imaging modalities to evaluate these lesions. The margins of the abscess are difficult to distinguish in the acute phase, and large amounts of cellular debris may result in a solid appearing mass, which is difficult to differentiate from a tumor. Historically, surgical exploration was employed to distinguish between renal abscess and renal carcinoma [11]. As the abscess coalesces, renal ultrasound will more reliably demonstrate a fluid-filled hypoechoic mass surrounded by a thick wall. Air within the abscess will produce a strong echo and posterior shadowing on ultrasonographic imaging. Angiography can also help differentiate abscess from tumor by demonstrating a hypovascular or avascular interior surrounded by a hypervascular rim, although this technique has the disadvantage of being more invasive [12].
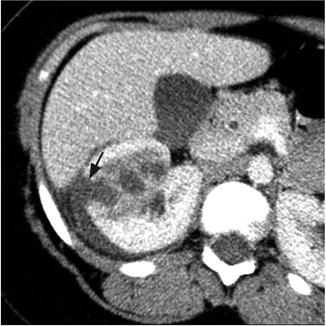
Fig. 3.2
Renal abscess. Contrast-enhanced CT of the right kidney shows area of poor perfusion in the right kidney and focal area of cortical disruption (black arrow) and crescentic pus collection around the right kidney
Given some of the limitations of ultrasound and angiography, CT is currently the most widely utilized imaging modality for this lesion, as it is noninvasive and provides the most accurate anatomic information [13]. In the acute phase of infection, CT may simply reveal an enlarged, enhancing mass, leading to an incorrect diagnosis of a neoplastic process [14]. Most abscesses will appear as low-density masses with rim enhancement; the presence of intralesional gas is pathognomonic for renal abscess [13]. Once the diagnosis is made, a variety of treatment approaches can be successful that include intravenous antibiotics alone in an abscess less than 3 cm or percutaneous drainage and/or surgical intervention for larger abscesses [15].
In the modern era, most renal abscesses are managed without surgical resection and are generally not seen in surgical pathology suites . However, renal abscesses may be seen at the time of autopsy and appear as yellow-brown infarcted regions within the renal cortex or medulla. An acute abscess may show the presence of purulent material on transection (Fig. 3.3).
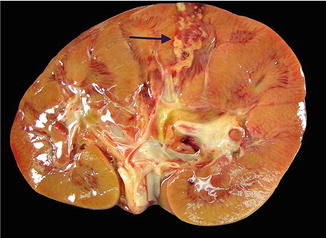
Fig. 3.3
Renal abscess on gross pathology . Gross image demonstrating the presence of a renal cortical abscess that is extending down to the medulla. The lesion grossly appears as a yellow, hemorrhagic area that may resemble a focus of infarction
Xanthogranulomatous Pyelonephritis (XGP)
XGP is an uncommon, chronic pyelonephritis resulting from an atypical, partial immune response to a subacute bacterial infection. XGP is usually seen in conjunction with renal obstruction and staghorn calculi in approximately 80 % of cases [16]. Common pathogens implicated include E. coli, P. mirabilis, S. aureus, Pseudomonas aeruginosa, and Klebsiella pneumoniae [17, 18]. XGP usually presents in middle-aged women, but can be seen in men. Diabetes is an established risk factor. The typical clinical presentation includes fever, chills, and flank or abdominal pain (Table 3.2). XGP may masquerade as a malignant process due to the frequent presence of nonspecific constitutional complaints including malaise, fatigue, and weight loss [19, 20]. As with other infectious processes involving the renal parenchyma, urinalysis is not specific. Blood work may reveal leukocytosis, anemia, abnormal liver function tests, hypoalbuminemia, hypergammaglobulinemia, and hyperuricosemia [19, 21].
Two distinct forms of XGP include the diffuse form in 85 % of cases and the localized form in 15 % of cases. Imaging studies commonly show nephromegaly, renal function impairment, and urinary obstruction secondary to calculi (Fig. 3.4) [22]. Ultrasound findings include diffuse renal enlargement, multiple hypoechoic areas, and echogenic foci with posterior acoustic shadowing suggesting calculi (Table 3.3). CT is the most useful imaging modality as it can define the extent of the inflammatory process and aid in surgical planning. Findings on CT that suggest XGP include renal enlargement, calcification of the renal pelvis, and multiple hypodense areas representing hydronephrotic calyces and abscess cavities (Table 3.3) [22, 23]. Contrast enhancement may additionally show wall enhancement, cortical thinning with reduced contrast excretion, and areas of fat attenuation due to lipid-rich xanthomatous tissue [23]. There are also rare case reports describing XGP associated with renal vein or caval thrombus and lymphadenopathy, further complicating the diagnostic picture [24, 25]. Given the more limited nature of localized XGP, a renal neoplasm is common in the differential diagnosis [26].
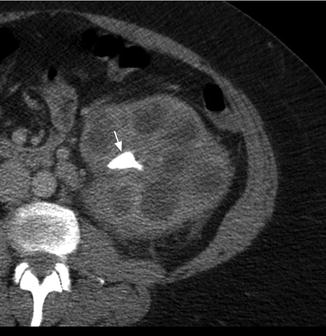
Fig. 3.4
Xanthogranulomatous pyelonephritis (XGP). Contrast-enhanced CT shows an enlarged left kidney with a staghorn calculus (white arrow) in the renal pelvis with multiple hypodense surrounding areas representing hydronephrotic calyces and abscess cavities
Table 3.3
Imaging findings suggestive of XGP
Ultrasound |
Diffuse renal enlargement |
Multiple hypoechoic masses |
Hyperechoic foci with posterior acoustic shadowing |
Hydronephrosis/pyonephrosis |
Perirenal/psoas abscess |
CT scan |
Diffuse renal enlargement |
Hydronephrosis/pyonephrosis |
Renal calculus |
Parenchymal thinning |
Nonfunctioning kidney |
Multiple hypodense areas |
Areas of fat accumulation |
Cutaneous fistula |
Abscess |
Suggest focal XGP |
Isolated pseudo-cystic mass |
Solitary calyceal dilation with calculus |
Adherent Gerota’s fascia |
Normal kidney function |
XGP reflects up to 19 % of the final diagnoses in cases of end-stage pyelonephritis [27] While nephrectomy remains the mainstay of treatment for XGP, optimal management likely includes appropriate antibiotic therapy, percutaneous drainage of associated abscesses, and possible percutaneous nephrostomy-tube drainage prior to surgical intervention [23]. While a laparoscopic approach may be attempted in cases of suspected XGP, it is associated with a high complication and conversion rate [27, 28]. Partial nephrectomy can be successful in cases of focal XGP [23, 29]. Additionally, if the diagnosis can be confirmed with percutaneous needle biopsy , there are rare reported cases of resolution of focal XGP with percutaneous drainage and antibiotics alone [30, 31].
On gross evaluation, the kidney is usually enlarged and shows a thickened capsule. The parenchymal surface may contain multiple, bright, yellow nodules. Necrosis of the renal parenchyma and dilatation of the renal pelvis secondary to renal calculi may also be seen. Microscopic sections show a granulomatous process (Fig. 3.5) that contains a mixed inflammatory infiltrate associated with a prominence of lipid-laden macrophages containing foamy cytoplasm (xanthoma cells) (Fig. 3.6). Surrounding fibrosis of the region may be seen. XGP may represent a diagnostic pitfall especially on frozen section evaluation, as foamy macrophages may be mistaken for clear cells associated with clear cell renal cell carcinoma. On permanent section, the presence of diffuse immunoreactivity for CD68 in the foamy macrophages and absence of immunoreactivity for epithelial markers in the lesion can aid in the diagnosis [32].
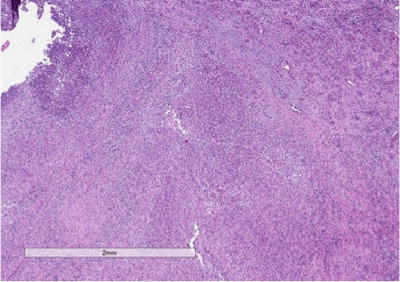
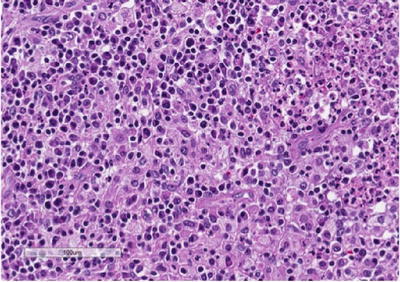
Fig. 3.5
Xantho-granulomatous pyelonephritis (XGP). Low-power view showing the architecture of the kidney being effaced by a mixed inflammatory infiltrate
Fig. 3.6
Xanthogranu-lomatous pyelonephritis (XGP) . High-power view showing a prominent mixed infiltrate composed of lipid-laden macrophages or so-called xanthoma cells, lymphocytes, neutrophils, and eosinophils. Xanthoma cells may represent a diagnostic challenge, especially on frozen section, and may be mistaken for clear cell renal cell carcinoma
Fine needle aspiration (FNA) may be used as an adjunct in preoperative diagnosis and will show xanthomatous cells with round vesicular nuclei that lack prominent nucleoli [32, 33]. The diagnosis on FNA can be especially challenging due to significant morphologic overlap with clear cell renal cell carcinoma. Immunohistochemistry can prove to be a useful adjunct in arriving at the correct diagnosis [34].
Renal Malakoplakia
Renal malakoplakia is a rare, chronic inflammatory disease that can affect any organ system but most commonly involves the urinary tract. While it shows a predilection for the urinary bladder, it may occur anywhere in the urinary tract including the upper tract and renal parenchyma. Malakoplakia has a predilection for immunocompromised patients and is also frequently seen in middle-aged women with a history of chronic urinary tract infections. Renal failure is also common at presentation [35]. Common symptoms include fever, dysuria, and urinary frequency and urgency [36, 37]. Gram-negative bacteria are often associated with malakoplakia, with E. coli and Proteus species accounting for the vast majority of cases [38]. The pathogenesis is thought to be secondary to an impaired host immune response to infection, likely abnormal macrophage function, that results in the formation of mass-like lesions in the urinary tract. While the process is usually multifocal, a single lesion can be seen in up to 25 % of cases [38]. Most cases with renal parenchymal involvement are unilateral, although there are reports of bilateral disease at this location [39].
The radiographic appearance of malakoplakia can vary and the findings are nonspecific. Malakoplakia may appear as a unilateral enlarged kidney due to diffuse infiltrative disease, as multiple parenchymal lesions, or as a solitary low-attenuation mass. Solitary lesions are particularly difficult to diagnose because they can mimic solid renal masses. Ultrasound may demonstrate a diffusely enlarged kidney with poorly defined hypoechoic masses and distorted renal architecture. CT scan may show multiple low-attenuation areas of various sizes, which may coalesce into a single larger mass [1, 40]. The diagnosis can be confirmed by endoscopic, percutaneous, or open biopsy. The treatment of malakoplakia varies depending on the extent of disease and the overall condition of the patient. Bilateral or multifocal disease can be treated with antibiotics with the addition of bethanechol, which helps correct the macrophage lysosomal defect. Surgical excision is the standard of care for unifocal disease [38].
Histology shows a granulomatous process associated with the presence of characteristic targetoid bodies called “Michaelis–Gutmann” bodies (Fig. 3.7). Michaelis–Gutmann bodies are thought to result from incompletely digested bacteria within phagolysosomes and the deposition of calcium and iron [41, 42]. Several stains including iron, calcium and PAS stains can highlight Michaelis–Gutmann bodies (Fig. 3.8, PAS shown).
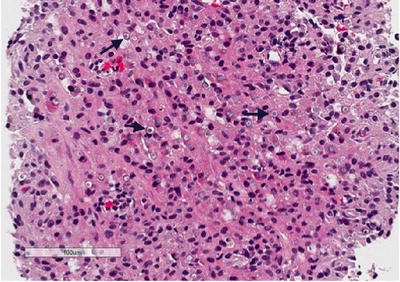
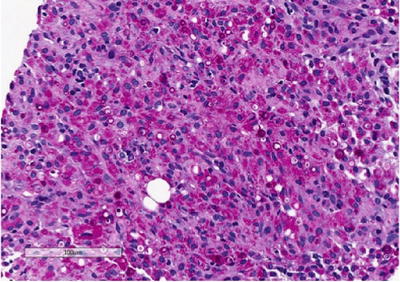
Fig. 3.7
Renal malakoplakia. Renal biopsy showing a granulomatous infiltrate in the background, with numerous targetoid “Michaelis-Gutmann” bodies (arrows)
Fig. 3.8
Renal malakoplakia. Periodic acid-Schiff (PAS) stain highlights Michaelis-Gutmann bodies that are believed to occur due to deposition of calcium and incompletely digested bacteria within phagolysosomes
Cystic Lesions of the Kidney
In this section, we will discuss autosomal dominant and recessive forms of polycystic kidney disease, as well as acquired renal cystic disease. Renal dysplasia, which may demonstrate cyst formation, is discussed in Chap. 11.
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
Autosomal dominant polycystic kidney disease (ADPKD) has an incidence of 1/500–1000 persons in the general population and results from a germline mutation in the PKD1 or PKD2 genes [43]. It is the most common inherited disease of the kidney and accounts for ~5 % of cases of end-stage renal disease (ESRD) in the United States [44]. ADPKD is a systemic disorder with clinical manifestations in many locations including the liver, seminal vesicles, pancreas, and intracranial circulation. Due to the autosomal dominant pattern of inheritance, there is a 50 % probability that the child of an affected parent will inherit the disease. The progressive loss of nephrons as a result of cyst formation usually does not become clinically significant until the third or fourth decade of life [45]. While most patients are asymptomatic, some may present early with abdominal or flank pain, hematuria, or urinary tract infections. Pain is the most frequent symptom of ADPKD, while hypertension is the most common sign of the disease [45, 46].
Patients with a family history of ADPKD should be screened after the age of 18 years; if ADPKD is diagnosed, the patient’s first-degree relatives should be screened as well [47]. ADPKD is diagnosed on imaging (Fig. 3.9) based on well-established criteria (Table 3.4) [48, 50]. Although CT and magnetic resonance imaging (MRI) are more sensitive for identification of renal cysts, ultrasound is the preferred imaging modality for screening as it is low cost and lacks radiation exposure. Genetic testing is not routinely recommended for ADPKD, but can be useful on a case-by-case basis [49].
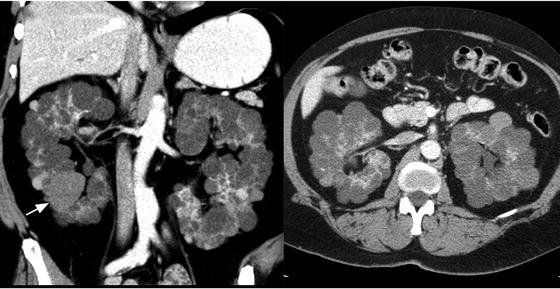
Fig. 3.9
Autosomal dominant polycystic kidney disease (ADPKD). Axial and coronal contrast-enhanced CT of the kidneys showing bilateral enlarged kidneys with the parenchyma replaced by multiple cysts, some that are hyperdense (white arrow)
Age (years) | Criteria |
|---|---|
Ultrasonography (if 50 % risk of disease)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |





