Indications
Contraindications
Relative
Absolute
Nephrotic syndrome
Severe azotemia
Uncooperative patient
Routinely in adults
Renal anatomic abnormalities
Uncontrollable hypertension
If atypical for MCD in children
Antiplatelet therapy or anticoagulation
Uncontrollable bleeding diathesis
Nephritic syndrome
Solitary native kidney
Systemic disease with renal insufficiency
Skin infection over the biopsy site
Unexplained CKD
Active kidney infection
Familial renal disease
Pregnancy
Renal transplant dysfunction
Percutaneous Kidney Biopsy
The percutaneous renal biopsy has been adopted as the principal technique for sampling renal parenchyma. When doing percutaneous biopsy, most renal pathologists agree that at least two core samples are necessary for adequate sampling and to avoid missed diagnoses. Where available, it is also useful to hand tissue directly to a trained technician at the time of biopsy for review under either a dissection or light microscope to evaluate for tissue adequacy based on the number of observed glomeruli [9].
This procedure is typically done with the patient awake and in the prone position. CT or ultrasound is used to locate the desired target—typically the lower pole of either kidney. The patient is given instructions on breath holding and regional anesthetics are used to numb the patient from the skin to the renal capsule. Since renal parenchyma lacks sensory innervation, there is no need to anesthetize deeper renal tissues. A biopsy needle is passed through the anesthetized tract and core samples taken with the patient instructed to hold their breath during period when the biopsy will be taken. Typically precautions are taken following the procedure to decrease the risk of bleeding and provide time for hemostasis to occur. Patients are maintained in the supine position with frequent vital sign monitoring for the ensuing 8–24 h. Urine collections are also preserved for physician review to ensure that gross hematuria, if present initially, improves during that same period.
A number of clinical studies have tried to address the exact amount of time that a patient should be observed for complications following the procedure. Early literature on the topic suggested that most complications would be noted by 12 h, thus perhaps eliminating the need for overnight hospitalization [10]. More recent data suggests that it may take longer to recognize complications, with one observational study noting that while 46 % of complications were noted before 4 h and 67 % by 8 h, 89 % could be identified by 24 h [11]. Presently, consensus recommendation is to observe patients in the inpatient setting for 24 h post procedure.
Open Kidney Biopsy
Laparoscopic and open biopsies have been shown to be largely safe and well tolerated. In the largest case series of 934 patients, a modified laparoscopic approach with general anesthesia had 100 % tissue adequacy for diagnosis, no major complications, and only one minor complication related to anesthesia [12].
However, given the invasive nature of the procedure, the risks of general anesthesia on a population basis, and the prolonged recovery time, open biopsy is typically only used in patients who have either an absolute or relative contraindication that precludes percutaneous biopsy [13].
Transvenous Kidney Biopsy
The transvenous—transjugular or transfemoral—approach is a procedural technique first described by Mal in France in 1990 [14]. Various reasons have been cited as prompting this choice of procedure, including need for other organ biopsies (heart, liver), bleeding diatheses, and uncontrolled hypertension, among others. Since that time a number of studies have been published reviewing the adequacy of this technique as well as their complication rates [13, 15]. Studies have reported adequate parenchymal sampling, with rates ranging from 73 to 97 %. In the published literature, no patient has died or required dialysis after these procedures [13]. Comparison to the other available biopsy options is difficult because of inherent selection bias. At present, the use of this technique largely depends on local availability and patient-specific factors that would preclude percutaneous biopsy.
Tissue Fixation and Preparation
The fixatives used for preserving the tissue vary by microscopy modality. It is important that the proceduralist be familiar with the handling preferences of their renal pathology lab in order to ensure appropriate handling once collected. Here we will discuss the handling of the percutaneous biopsy, given its prevalent use in parenchymal sampling.
The core samples taken by percutaneous biopsy are typically divided into portions destined for the varying histologic uses. Samples are taken from both the cortical end of the tissue and the medullary end for use in electron microscopy. The remaining tissue from each tissue core is divided for use with immunofluorescence of light microscopy.
Light Microscopy
Most pathology laboratories prefer light samples to be collected and stored in 10 % aqueous formaldehyde solution (formalin). Formalin is safe and readily disposed of and preserves morphologic features for review. It also allows for additional tissue processing using immunohistochemical techniques or molecular studies to be used if necessary. Alternative solutions include Bouin’s, Duboscq-Brazil, Zenker’s, or 4 % paraformaldehyde fixatives, each with varying uses [9].
Slide preparation also varies according to pathology laboratory, but typically includes sectioning of the preserved tissue into 2 μm slices and serial staining with hematoxylin and eosin (H&E) stain , periodic acid-Schiff reaction (PAS), silver stain, or trichrome stain [9].
Electron Microscopy
Tissue fixation for EM requires fixation in either 2–3 % glutaraldehyde or 1–4 % paraformaldehyde. This provides the greatest degree of structural preservation. It is occasionally necessary to reprocess samples stored in either the frozen section or the paraffin-embedded section if glomeruli are not available on the EM processed sections. This can lead to cellular artifact, but generally is considered acceptable for review of the GBM for both structural integrity and for immune deposition [9].
Immunofluorescence or Immunoperoxidase
Some pathology laboratories have gone to immunoperoxidase (IP) staining for review of immune deposition due to relative ease of processing. However, most renal pathologists agree that dark field imaging with IF is the preferred modality to identify immune deposition in pathology samples. The processing of these materials is laboratory dependent, though formalin fixation is typically adequate with freezing and subsequent sectioning used to label the immune complexes [9].
Glomerular Syndromes and Mechanisms of Injury
Medical renal disease affects the kidneys in a variety of ways and can manifest clinically as any number of clinical syndromes. Each of the individual classes of renal disease has their own hallmarks; here, however, we will be focusing diseases primarily affecting the glomerulus.
The glomerular diseases manifest according to the pattern of underlying injury, which we will briefly review, before discussing specific clinical syndromes.
Structural Patterns of Injury
As discussed previously, the glomerular tuft houses the filtration apparatus that generates the tubular ultrafiltrate and is composed of three layers: the fenestrated endothelium, the glomerular basement membrane, and the podocytes. Each of these layers can suffer injury and the nature of that injury correlates closely with the observed clinical syndrome.
Immune conditions or vasculitis conditions that affect the endothelium lead directly to a robust inflammatory cascade that typically leads to endothelial proliferation and filtration barrier disruption. That disruption can be detected clinically as the nephritic syndrome with loss of both protein and cellular elements (typically red blood cells) into the urinary space with concomitant changes in filtration efficacy as detected by changes in the glomerular filtration rate (GFR).
Conditions that affect the glomerular basement membrane include both congenital and acquired conditions. The congenital disorders tend to be more indolent with adaptive changes in the associated glomerular cell types and long latency to diagnosis. These include disease like thin basement membrane disease and Alport’s disease . The acquired types can lead directly to disruption of the GBM and has implications for both the size and charge selectivity of the filtration barrier. These acquired conditions, like anti-GBM disease, typically lead to robust immune activation and consequences similar to those seen with endothelial diseases.
The podocytopathies are a family of disease that classically disrupt the cellular architecture of the podocyte leading to breakdown of the interdigitated foot processes with associated loss of charge selectivity. That loss of charge specificity leads to robust proteinuria, but relatively limited, if any, hematuria.
Each of these will be discussed in more detail below.
Nephritic Syndromes
The nephritic syndrome is a collection of signs and symptoms that denote underlying glomerular injury, the hallmark of which is the presence of both proteinuria and hematuria in the urine. The classic nomenclature is used to describe a limited amount of protein, with any amount of glomerular hematuria. This contrasts with the nephrotic syndrome, which denotes a disease wherein there is significant proteinuria and no hematuria. Some providers also discuss the nephritic/nephrotic syndrome where there are significant hematuria and nephrotic-range proteinuria, though this is better described as an advanced nephritic syndrome, given that the underlying mechanism of injury, as discussed above, mandates endothelial injury in order to facilitate the observed hematuria.
Rapidly Progressive Glomerulonephritis
The most striking examples of the nephritic syndrome are the crescentic glomerulopathies , which frequently present as rapidly progressive glomerulonephritis (RPGN). RPGN does not refer to a single disease process; rather, it describes a clinical syndrome of rapidly progressive acute renal failure associated with hematuria and proteinuria, which is typically caused by advanced histologic injury to the glomerular tuft.
Clinical Presentation
Clinically, this collection of diseases share certain features, namely, the presence of a nephritic sediment with red blood cells either in red cell casts or with dysmorphic characteristics, some amount of proteinuria, and rapid loss of renal function. Renal failure with oliguria, severe azotemia, and hypertension are the hallmark of the initial presentation and typically lead to progression to ESRD within 3 months of diagnosis if left untreated [16]. Patients may also have evidence of hypertension—owing to disrupted sodium regulation and possibly renin-mediated mechanisms—and edema. There are of course more distant or systemic effects that can be seen as well, though these are generally a characteristic of the underlying disease. An example would be the pulmonary hemorrhage seen in anti-GBM disease (Goodpasture’s disease) [17].
The number of disease conditions that are known to cause RPGN is limited. Each has a specific pathogenic mechanism by which it leads to glomerular capillary injury and disruption, outlined separately, but they can be organized according to their pattern of injury. The first and most straightforward is anti-GBM disease, the second group being a far more heterogeneous array of disease wherein immune complexes in the glomerular capillary tuft lead to local injury, and the last being the pauci-immune vasculitides wherein no immune depositions can be detected, but pathogenic antineutrophil cytoplasmic antibodies (ANCA) lead to notable inflammatory activation which can be observed histologically. The latter is the most common form of crescentic glomerulonephritis , accounting for approximately 60–80 % of all cases [16].
Pathogenesis
While the underlying pathogenesis of the specific disease entities is reviewed below, their progression to an RPGN is typical and includes necrotizing inflammation which leads to GBM disruption and leakage of serum proteins including fibrin and fibronectin into the urinary space which generates a robust activation of both the visceral and parietal epithelial layers in Bowman’s space. In turn, these cells dedifferentiate and proliferate and lead to the formation of the prototypical crescent (Fig. 12.1) [16].
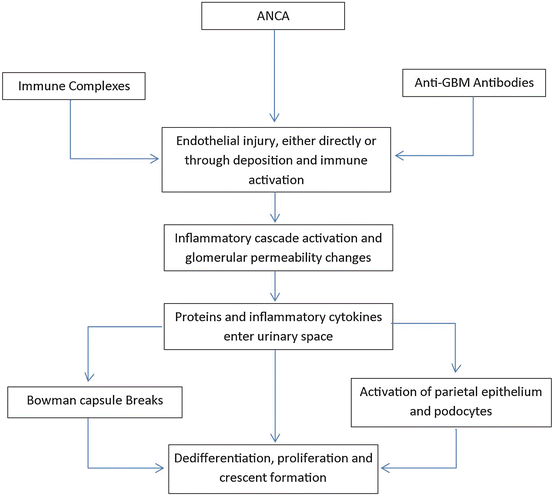
Fig. 12.1
Pathogenesis of crescent formation
The crescents themselves are a histologic signal of injury; the association of this pattern of injury with the clinical manifestations of the RPGNs has led to these terms being considered near synonymous [18].
Treatment
Despite their differences, the initial treatment of these diseases is similar, largely because of the incredibly high risk of irreversible renal injury and risk of other morbidities and mortality [19]. Typically the treatment of any of these diseases is divided into induction and maintenance phases. Induction therapy is often initiated in advance of definitive diagnosis. Sometimes this is with serologic evidence, but often, even that is not yet available. The cornerstones of treatment have been steroid therapy and plasma exchange for nearly 40 years [20]. Recent meta-analysis on the topic has shown that the use of adjunct plasma exchange can reduce the rate of progression to ESRD in these patients, noting that ESRD rates are reduced at both 3 and 12 months [21]. Pulse glucocorticoid therapy is also typically started at the time of clinical presentation. While subject to varying patterns of administration, typically 500–1000 mg of methylprednisolone is given over 2–3 days as initial therapy to help calm the inflammatory cascades active in the tuft. Lastly the use of immunosuppression therapy is used to reign in the production of the injurious autoantibodies. The classical treatment for RPGN has been oral or intravenous cyclophosphamide (CYC) administered according to a variety of protocols either as continuous oral therapy or as pulse intravenous therapy [21]. With the advent of newer immunosuppressive therapies, the treatments used in practice have increased, though the details of their selection are outside the scope of this publication.
Anti-glomerular Basement Membrane Disease
Typically this disease presents as a pulmonary renal syndrome with acute onset of both lower respiratory complaints and changes in urinary character [17, 19]; however, more subtle cases can be seen. Because of its tendency to rapidly progress, it should be considered in all cases of acute pulmonary renal disease, and coordination with a nephrologist is recommended for urgent initiation of plasmapheresis if needed in the setting of diffuse alveolar hemorrhage [21, 22].
Pathogenesis
The GBM is formed through the fusion of the endothelial and podocyte basement membranes. The normal GBM is a tight meshwork of collagen fibers, with alpha 3, alpha 4, and alpha 5 subtypes of type IV collagen fibers forming hexamers that then crosslink through their non-collagenous (NC1) domains. In patients with anti-GBM disease, immunoglobulin G (IgG) can be identified on immunofluorescence overlying all of the glomerular basement membrane, and the causative antibodies have been found to be directed against those NC1 domains of alpha 3 or alpha 5 type IV collagen [23].
Recent work has demonstrated that the disease is one of quaternary conformational change—the specific cause of which is unknown, though it has been linked to smoking, viral infections, and solvent exposure—which leads to molecular conformational shifting and exposure of the normally sheltered NC1 domains with subsequent autoantibody formation. Those autoantibodies then bind to the NC1 domains and lead complement fixation and to a type II hypersensitivity reaction with robust inflammatory recruitment and consequent local injury [22, 23].
This disease manifests as the nephritic syndrome due to the ongoing glomerular injury, and patients have pulmonary symptoms as well given that the alpha-3 subunit is also expressed in the alveoli of the lungs. This can lead to a very dramatic clinical presentation of the disease with diffuse pulmonary hemorrhage as the initial presenting symptom. Prior to the use of the above-described therapies, anti-GBM disease was nearly universally fatal, though with the use of modern treatments, the 5-year survival now exceeds 80 % [22].
Pathology
Anti-GBM disease has characteristics on light microscopy that are typical for RPGN, typically with areas of GBM destruction with nearby fibrinoid necrosis. In early disease, there may not be evidence of crescent formation on the biopsy sample, though in more advanced cases, lymphoplasmacytic infiltration and endocapillary proliferation may become apparent. Immunofluorescence is diagnostic with smooth GBM staining for IgG and c3. IgG is usually more positive than c3 staining. Electron microscopy reveals the absence of immune deposits and can highlight the broken GBM with associated fibrin tactoids notable in areas of discrete necrosis.
Management
The challenges present in diagnosing this disease combined with its rarity make this a challenging disease on which to do randomized clinical trials. However, given that it is understood to be an immune-mediated disease, immunosuppressive therapies are considered necessary as part of its treatment. If diagnosed during its fulminant phase, plasmapheresis is often indicated. At the same time, induction is achieved, as described above for all of the RPGNs, classically done with CYC and high-dose glucocorticoid therapy [16, 19, 22]. The introduction of anti-CD20 monoclonal antibody (rituximab) therapies has been used as well in small patient cohorts, the largest a series of eight patients who had received steroids and CYC as well as plasmapheresis. All were started on rituximab within 2 months of diagnosis. At approximately 2 years follow-up, patient and renal survival were 100 and 75 %, respectively [24].
The prognosis for patients affected by anti-GBM disease is largely dependent on the promptness of the diagnosis and initiation of effective treatment. While historically the disease was considered fatal [25], the above therapies have led to a 5-year survival rate in excess of 80 %, and only about a third of patients require long-term dialysis [22].
Pauci-immune Glomerular Disease
Accounting for just over half of all diagnosed RPGN syndromes and sometimes recognized in more indolent circumstances, this group of diseases is characterized by necrotizing changes on biopsy in the absence of immune deposits [19]. This group of diseases includes granulomatosis with polyangiitis (GPA), eosinophilic granulomatosis with polyangiitis (EGPA , formerly Churg-Strauss syndrome), and microscopic polyangiitis (MPA). The bulk of these patients have circulating ANCA. Clinically they present in a variety of different ways, each of which is typically associated with a collection of extrarenal manifestations that can suggest which disease is causative, though it does require tissue biopsy to establish the definitive diagnosis. The varying extrarenal characteristics, as well as the frequency of specific ANCA subtype s with those diseases, are summarized in Table 12.2. These diseases do occasionally present with less fulminant disease. So-called early systemic disease is defined as serologic positivity, but with sCr below 1.7 mg/dL and no critical extrarenal organ dysfunction [26]. This distinction has implications for treatment.
Table 12.2
Features of ANCA-associated vasculitides
Renal limited disease | Microscopic polyangiitis | Granulomatosis with polyangiitis | Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) | |
|---|---|---|---|---|
Frequency of renal involvement | 100 | 90 | 80 | 45 |
Frequency of ANCA positivity | 80 | 90 | 95 | 70 |
MPO | 60 | 50 | 25 | 60 |
PR3 | 40 | 50 | 75 | 40 |
Extrarenal manifestations | None | Musculoskeletal complaints, pulmonary | Pulmonary, sinus, head, and neck | Pulmonary, neurologic, allergy symptoms |
Pathogenesis
The association of ANCA and necrotizing glomerular disease was first recognized by Davies in 1982 [27]; subsequent work has gone on to support a pathogenic mechanism for these autoantibodies. There are two identified culpable antibody subtypes, the anti-myeloperoxidase (MPO) subtype and the anti-proteinase 3 subtype (PR-3). These antigens are typically sequestered into neutrophilic granules and are exposed only during periods of inflammatory activation [17, 19]. Directly linking this antibody activity to the disease, however, has been proven difficult. Renal biopsies of affected patients are rife with activated neutrophils, and indeed, the number of activated neutrophils seen on biopsy has been correlated with the degree of renal insufficiency [28], but they do not have observable immune complexes attributable to ANCA, hence the name “pauci-immune.” Additional evidence for a pathogenic role of ANCA has grown with ANCA being elutable from affected kidney tissue, and in the case of anti-MPO antibodies, necrotizing glomerulonephritis can be induced with infusion of ANCA [29]. Interestingly, anti-LAMP 2 (antibodies directed against lysosomal membrane protein-2) have been shown to be present in upward of 80 % of all active ANCA-associated vasculitides (AAV) [30, 31]. While the understanding of the disease is improving, there are many aspects of the pathogenesis that remain to be understood.
Pathology
The ANCA-associated vasculitides have similar histologic appearances. Common among all of them are segmental glomerular necrosis with crescent formation (Fig. 12.2) and the absence of either immunofluorescence findings or changes by electron microscopy. In GPA and microscopic polyangiitis, there is typically evidence of an active interstitial nephritis with or without larger vessel involvement. In EGPA there may or may not be an eosinophilic predominance in the interstitial infiltrate, and this does not have bearing on the pathologic diagnosis. The granulomatous changes seen in GPA are more typically identified in lung biopsy than in renal biopsy, and often the diagnostic distinction between these diseases (GPA/MPA/EGPA) is made clinically.
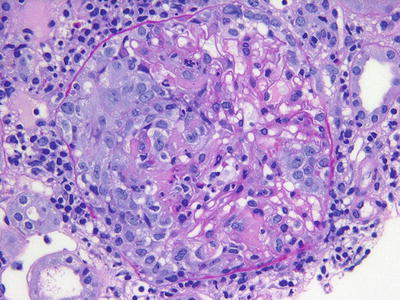
Fig. 12.2
A cellular crescent with fibrinoid necrosis is noted on the left side of this glomerulus in a patient with pauci-immune (ANCA-associated) glomerulonephritis (PAS)
Management
Again, the induction therapy for this disease has included treatment as described for an RPGN with cyclophosphamide as the core of the induction immunosuppressive regimen since the 1970s. Typically, maintenance therapy is necessary due to high rates of relapse and has historically been accomplished with azathioprine. Recent studies have also set out to explore the utility of B cell-depleting agents like rituximab both as an alternative induction agent and a maintenance therapy.
In situations where patients may present with less threatening disease, so-called early systemic AAV, several studies have shown non-inferiority of methotrexate (MTX) as an induction agent. In one randomized controlled trial patients with confirmed ANCA positivity, sCr <1.7 mg/dL and no evidence of critical organ dysfunction were randomized to receive either oral cyclophosphamide or oral MTX for 12 months. There were similar rates of remission at 18-month follow-up, though the time to remission was longer in the MTX patients, particularly those with more extensive disease [26].
Cyclophosphamide is the historical gold standard for induction therapy. Oral or intravenous (IV) administration of CYC has also been extensively studied [32–34]. Indeed similar response rates were seen in patients receiving either oral or IV CYC, though the cumulative dose of those patients receiving IV therapy was less. Given that the complication profile of CYC is largely directed by its cumulative dose, there may be some long-term benefit to limiting the absolute amount of CYC administered. However, long-term follow-up of patients previously enrolled in the largest of these trials has failed to show a difference in treatment-related adverse events. It did, however, show a higher relapse rate among patients receiving the IV therapy, though this ultimately was not reflected in differences in the overall patient morbidity or mortality at just over 4 years [35].
Rituximab has also been used for both induction and maintenance in these patients as well with high efficacy. Two large, well-conducted trials have evaluated the use of rituximab for induction therapy with similar efficacy [36, 37]. Follow-up trials have gone on to show non-inferiority for maintenance therapy with rituximab as well [38]. The current treatment recommendations for AAV are summarized in Table 12.3.
Table 12.3
Overview of the treatment of AAV according to EULAR recommendations
Disease stage | Treatment | Study/level of evidence, grade of recommendation |
|---|---|---|
Induction of remission | ||
Early systemic | Methotrexate 15 mg/week (oral/parenteral), increase to 20–25 mg/week, folic acid + GC | NORAM (level 1B, grade B) |
Generalized | Cyclophosphamide IV/oral + GC | CYCLOPS (level 1A/1B, grade A) |
duration: 3–6 months (oral) or 6–9 pulses (IV) | RAVE (level 1B) | |
Rituximab 4 × 375 mg/m2 in weekly intervals | ||
Severe (creatinine >500 μmol/L) | Standard therapy + plasma exchange | MEPEX (level 1B, grade A) |
Rituximab 4 × 375 mg/m2 in weekly intervals (as substitute for cyclophosphamide) | RITUXVAS (level 1B) | |
Concomitant glucocorticoids (GC) | Prednisolone/prednisone 1 mg/kg/day oral taper to 15 mg/day or less within 3 months | (level 3, grade C) |
Maintenance of remission | ||
Maintenance options | Azathioprine 2 mg/kg/day oral + low-dose GC | CYCAZAREM (level 1B, grade A) |
Methotrexate 20–25 mg/week + low-dose GC | WEGENT (level1B, grade B/A) | |
Leflunomide 20 mg/day oral + low–dose GC | LEM (level 1B, grade B) | |
Duration: at least 18 months | ||
Concomitant GC | Prednisolone/prednisone less than 10 mg/day
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |





