Fig. 9.1
Mucinous and tubular spindle cell carcinoma. (a) Inter-anastomosing cords of cells in a mucinous background are seen at low magnification. (b) Tumor cells show relatively uniform nuclei with minimal atypia
MTSCC has been shown to harbor multiple chromosomal alterations , including 1, 4, 8, 9, 13, 14, 15, and 22 [16]. The genetic changes characteristic of papillary RCC, namely, trisomy 7 and 17 and loss of the Y chromosome, are not identified in MTSCC [17].
Most MTSCC lesions express markers that are reminiscent of distal nephron differentiation, including EMA, AE1/3, CK7, E-cadherin, and α-methylacyl CoA racemase. RCC antigen is positive in the majority of cases. The proximal tubular marker CD10 is positive in only 10–15 % of MTSCC cases [18, 19]. The immunohistochemical profile of MTSCC is nonspecific and partially overlaps with papillary RCC with the absence of CD10 being the most supportive immunohistochemical biomarker for the former [6].
Prognosis and Clinical Management
MTSCC is often indolent and surgery is curative in most cases [20–22]. A small subset of cases may recur or be associated with regional lymph node metastases [13, 16, 22, 23]. Sarcomatoid transformation is associated with an increased likelihood of distant metastases and risk of death from disease [14, 24, 25].
Clear Cell Papillary RCC
Introduction
Clear cell papillary RCC (also referred to by some as “clear cell tubulopapillary RCC ”) is reported to account for less than 5 % of renal epithelial neoplasms ; however, this incidence is likely an underestimation due to the misclassification of this tumor as a clear cell or less frequently a papillary, RCC. Clear cell papillary RCC affects men and women equally, and the age distribution ranges from 18 to 88 years [26–28]. Clear cell papillary RCC may occur sporadically or may be associated with end-stage renal disease [29–33].
Clinical Presentation
The majority of clear cell papillary RCC cases are discovered incidentally, although a subset of patients may present with hematuria or flank pain [27].
Pathology
Clear cell papillary RCC is primarily a cortical lesion . On gross evaluation, these tumors are well circumscribed, encapsulated, and relatively small in size (0.6–5.5 cm) [28, 31]. Cystic change may be present. The cut surface ranges from yellow to tan to brown in appearance. These tumors are commonly unifocal, although bilateral and multifocal tumors have been described.
Microscopically, several architectural patterns may be present and include cystic and solid structures as well as branching glands [27, 31, 34]. A defining characteristic of these tumors is the presence of tubular and papillary structures lined by clear cells (Fig. 9.2a). Occasional eosinophilic luminal secretions may be seen. Tumor cells show clear cytoplasm, and the nuclei are uniform and small and lack nucleoli (Fig. 9.2b). Prominent nuclear polarization toward the luminal surface is striking in these cases [27, 31, 35]. The intervening stroma is often fibrous or hyalinized but may contain smooth muscle.
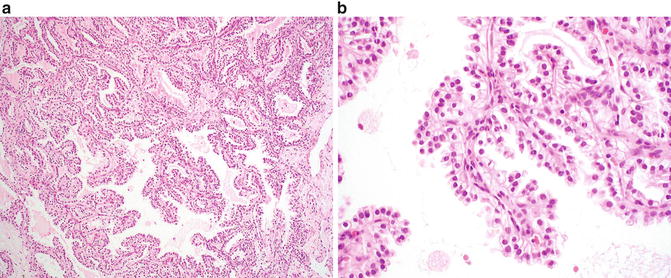
Fig. 9.2
Clear cell papillary renal cell carcinoma. (a) A combination of papillary and nested patterns is identifiable in this example. Uniform alignment of the nuclei to the luminal surface is evident even at low magnification. (b) Papillary fronds lined by clear cells are a characteristic feature of this lesion
Genetic analysis shows these tumors to be distinct from clear cell RCC and papillary RCC. Specifically, clear cell papillary RCC lacks chromosome 3p deletions or VHL gene alteration characteristic of clear cell RCC, as well as extra copies of chromosomes 7 and 17 as is seen in papillary RCC [30, 31, 36–39]. Some molecular differences have been reported between clear cell papillary RCC that occurs in the sporadic and the ESRD settings [31, 40, 41].
Immunohistochemistry is helpful in the diagnosis of this entity by distinguishing it from both clear cell RCC and papillary RCC. Clear cell papillary RCC shows positive immunoreactivity for CK7; carbonic anhydrase IX (CAIX), PAX2, and PAX8; and high-molecular-weight cytokeratin (34βE12) [26, 29, 42]. Immunostains for AMACR (racemase) and RCC antigen are negative, and CD10 is negative or only focally positive. In contrast, clear cell RCC is typically negative for CK7 and positive for CD10, RCC antigen, and CAIX and may show variable AMACR staining. Papillary RCC is typically positive for CK7, AMACR, and CD10; RCC antigen may be variable and CAIX should be negative.
Tubulocystic Carcinoma
Introduction
Tubulocystic carcinoma is an uncommon form of RCC described in numerous case reports and small case series [6, 43–46]. Tubulocystic carcinoma was initially described by Amin et al. [43] in 2009; however, earlier descriptions of similar tumors date back to1956, when it was included with Bellini (collecting duct) carcinomas, and later described as the low-grade collecting duct carcinoma [47, 48]. However, more recent studies have demonstrated that tubulocystic carcinomas are more likely related to papillary RCC instead of collecting duct carcinoma [43, 44]. Tubulocystic carcinoma affects adults over a broad age range of 29–94 years of age [49, 50]. There is a strong male predominance (male-to-female ratio of 7:1) [47, 51]. Based on immunohistochemical staining pattern, ultrastructural features, and gene expression profiling, tubulocystic carcinoma is favored to originate from either the proximal convoluted tubule or intercalated tubule [6, 43, 51].
Clinical Presentation
Patients are often asymptomatic, although some may present with abdominal pain, distension, and hematuria. Tubulocystic carcinoma is usually solitary and often involves the left kidney [51]; however, multifocal tumors have been described in approximately 23 % of patients [44, 47]. Tubulocystic carcinoma should be considered in the differential diagnosis of a renal lesion with a cystic component on radiological examination.
Pathology
On gross examination, tubulocystic carcinoma is usually solitary and well circumscribed and has a tan-gray, spongy appearance; the latter is due to numerous cysts/microcysts [47]. Tubulocystic carcinoma varies in size from 0.2 to 17 cm [48, 51]. Multifocal tumors may be present, which can include multiple distinct tubulocystic carcinomas or tubulocystic carcinoma associated with a concurrent second renal neoplasm, most frequently a papillary RCC [47, 48]. Tumors are most commonly located in the cortex, although up to a third can be present at the corticomedullary junction [6].
Microscopically, tubulocystic carcinoma is composed of many cystic structures that are small to medium in size, which are separated by thin fibrous septa (Fig. 9.3a). Solid growth is not seen. The cells lining the cysts are cuboidal, columnar, or hobnail in appearance with abundant eosinophilic or amphophilic cytoplasm which characteristically contain large nuclei with prominent red nucleoli (Fig. 9.3b) [47]. Rarely, sarcomatoid features may be seen in association with this tumor but only in the presence of solid growth (i.e., an associated component of high-grade papillary RCC) [1, 52]. On ultrastructural examination, the tumors show abundant microvilli with brush border organization suggestive of proximal convoluted tubules, with some features resembling intercalated cells of the collecting ducts [51].
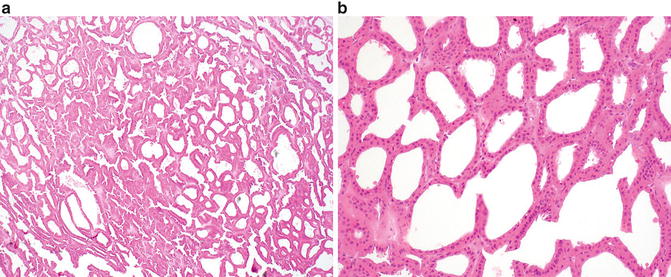
Fig. 9.3
Tubulocystic renal cell carcinoma. (a) Multiple cystic structures of variable size are present at low magnification. (b) Cysts are lined by a single layer of cells with eosinophilic cytoplasm , large nuclei, and prominent nucleoli
Genetic analysis of these tumors report a similar profile to papillary RCC, which include gains of chromosome 7 and 17 and loss of the Y chromosome. However, classic features associated with “low-grade” papillary RCC are typically not seen in this entity; in contrast, high-grade tumors with solid and papillary growth patterns and similar nuclear and immunophenotypic features can be seen [44, 45, 53]
Immunohistochemical studies demonstrate immunoreactivity for cytokeratins 8, 18, and 19 [6, 44]. CD10 and P504S (racemase) are positive in greater than 90 % of cases. Diffuse and strong AMACR positivity, positive to variable CK7 expression, and staining for kidney-specific cadherin and PAX2 have been reported [44, 47]. Tubulocystic carcinoma is negative for high-molecular-weight cytokeratin (34BE12) [47, 48].
The main differential diagnoses include other tumors with a multiloculated gross appearance , including multiloculated clear cell RCC, cystic clear cell tubulopapillary RCC, cystic nephroma, mixed epithelial and stromal tumor, and cystic oncocytoma [47, 51, 54, 55]. Distinction between these entities is primarily based on light microscopic evaluation, rather than use of ancillary studies.
Prognosis and Clinical Management
Whereas the majority of reported cases of tubulocystic carcinoma demonstrate indolent behavior, recurrence and metastases to regional and distant sites such as the bone and liver have been reported in rare cases; the latter is associated with tumors that demonstrate solid and papillary growth patterns [44, 45, 47, 51, 56].
Acquired Cystic Disease-Associated RCC
Introduction
Cystic change occurs in the majority of patients with end-stage renal disease (ESRD) who undergo dialysis and is termed “acquired renal cystic disease” (ARCD) [5, 6]. Patients with ARCD have increased risk of developing RCC comparing to normal population [6, 57, 58], with an RCC incidence rate of 10 % in ARCD patients [5, 6, 37, 59]. A direct correlation between the duration of dialysis and RCC development in ESRD patients has been suggested in some studies [5, 6, 29, 60]. Multiple types of renal neoplasms may arise in the background of ESRD, including clear cell RCC, papillary RCC, clear cell papillary RCC, and chromophobe RCC [5, 61–63], although acquired cystic disease-associated RCC represents up to 36 % of all renal neoplasms in this setting [29]. Similar to non-ESRD RCC, acquired cystic disease-associated RCC occurs most commonly in men between the ages of 55 and 65 years [5, 30, 40, 61].
Clinical Presentation
Pathology
On gross examination, acquired cystic disease-associated RCC is well circumscribed and tan/yellow in appearance and may show focal hemorrhage or necrosis [5, 6]. The background renal parenchyma shows multiple variably sized cysts, with the neoplasm appearing to arise in association with a cyst wall or from one of the connecting trabecular areas. Multifocal tumors occur approximately 50 % of cases, and bilateral tumors are found in up to 20 % of patients [1, 29].
At low magnification, acquired cystic disease-associated RCC shows a distinct “sieve-like” appearance (Fig. 9.4a). At higher magnification, multiple growth patterns that include acinar, alveolar, solid, cystic, and papillary regions may be identified. The tumor cells show granular, eosinophilic cytoplasm and contain large nuclei with prominent nucleoli [6]. Intratumoral calcium oxalate crystal is a relatively specific feature in this entity, although may be lacking a minority of cases (Fig. 9.4b) [6, 29, 59]. Rhabdoid and sarcomatoid features have been reported in a small number of cases [29, 61, 64].
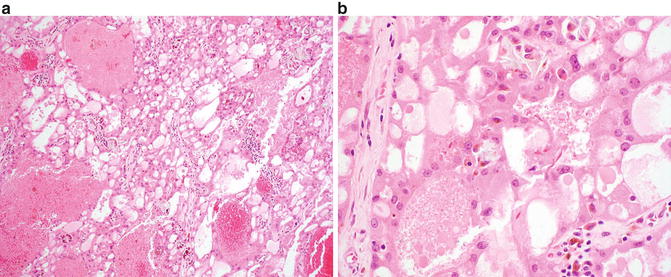
Fig. 9.4
Acquired cystic disease-associated renal cell carcinoma. (a) A sieve-like pattern is evident at low magnification. (b) Crystals may be seen in a subset of cases
Fluorescence in situ hybridization (FISH) and comparative genomic hybridization performed on nine cases showed variable combined gains of chromosomes 3, 7, 16, 17, and Y [6, 60].
Acquired cystic disease-associated RCC shows immunoreactivity for a-methylacyl-coenzyme A racemase (AMACR), CD10, RCC antigen, and glutathione S-transferase A [6, 60]. CK7 is often negative but can show focal immunoreactivity [6].
The main differential diagnosis includes papillary renal cell carcinoma. Immunostains for CK7 and chromosomal analysis can be used to distinguish these two entities in most cases, if required.
Prognosis and Clinical Management
Most cases of acquired cystic renal disease-associated RCC typically show indolent behavior, which may reflect early detection in patients with ESRD [6, 62]. As anticipated, the minority of tumors that show rhabdoid or sarcomatoid features may behave more aggressively and be associated with metastatic spread [1, 29, 64, 65].
Neuroblastoma-Associated RCC
Introduction
Neuroblastoma-associated RCC is a unique entity that occurs 3–35 years after diagnosis and treatment of childhood or adult neuroblastoma [5, 6, 66, 67]. It is a rare tumor, accounting for less than 1 % of all cases of RCC and approximately 2.5 % of the cases of RCC in children [5, 11]. Males and females have an equal risk of involvement [63]. Some reports suggest a relationship between the treatment for neuroblastoma and development of neuroblastoma-associated RCC [11]. However, a small number of patients develop RCC in the absence of radiation or chemotherapy, suggesting a possible underlying germ line susceptibility for these lesions [67].
Clinical Presentation
Pathology
On gross evaluation, tumors range in size from 1 to 8 cm [5, 63]. Neuroblastoma-associated RCC is histologically heterogeneous [11]. The most common appearance is that of an oncocytic tumor that shows solid and papillary architecture and occasional vacuolization of the cytoplasm [5, 53, 63, 68]. Variable nuclear size and medium-sized nucleoli are present, and mitotic figures may be identified. One should note that other forms of RCC, including Xp11.2 translocation-associated carcinoma and classic clear cell RCC, can also occur in a subset of patients following neuroblastoma [5, 63, 69].
Due to the small number of cases described to date, the genetic profile of this tumor has not been well defined, although chromosomal alterations affecting 14q31 and 20q13 have been reported [5, 70].
Immunohistochemistry shows neuroblastoma-associated RCC to be frequently positive for EMA, vimentin, and cytokeratins 8, 18, and 20 and negative for cytokeratins 7, 14, 19, 20, S100, and HMB45 [5, 63]. The main differential diagnosis includes oncocytoma and chromophobe RCC in the oncocytic subtype of neuroblastoma-associated RCC. Clinical history is critical in this differential diagnosis.
Prognosis and Clinical Management
The oncocytic subtype of neuroblastoma-associated RCC behaves in an indolent manner. Outcomes associated with clear cell RCC and Xp11-associated RCC are likely similar to their de novo counterparts. Awareness of this entity is important to ensure close follow-up and frequent assessment of survivors of neuroblastoma in order to detect RCC early in the course of disease.
Thyroid-Like Follicular Carcinoma of the Kidney (TLFCK)
Introduction
Thyroid-like follicular carcinoma of the kidney (TLFCK) is a rare renal neoplasm. To date, only a limited number of TLFCK cases have been reported in the literature [1, 6, 71–75]. This tumor occurs over a broad range of 29–83 years, with an equal gender distribution [1, 6, 73, 75]. This entity has not yet been classified as a distinct renal neoplasm by the World Health Organization.
Clinical Presentation
Pathology
On gross examination, these tumors are well defined and encapsulated, with a solid or cystic and yellow-tan cut surface [71, 75]. Focal areas of hemorrhage or necrosis may be seen. Microscopically, TLFCK shows variably sized follicular structures containing both microfollicular and macrofollicular patterns (Fig. 9.5a). The follicles contain pink colloid-like material and are lined by a single layer of cuboidal to columnar cells with moderate amphophilic to eosinophilic cytoplasm (Fig. 9.5b) [71, 73]. Nuclei are typically round to oval and may demonstrate nuclear grooves, similar to that seen in thyroid neoplasms. Lymphocytic infiltrates with or without reactive germinal centers can be present [71].
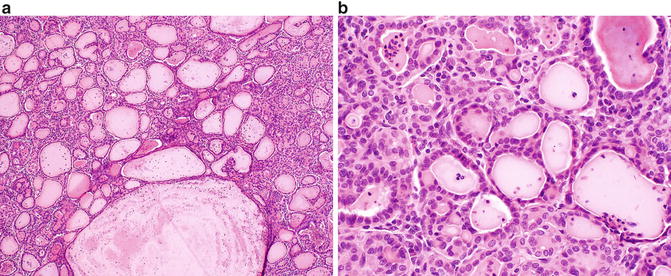
Fig. 9.5
Thyroid-like follicular carcinoma of the kidney. (a) A combination of variably sized follicular structures filled with colloid-like material and small nests may be seen in this entity. (b) The follicular structures are lined by a single layer of cuboidal to columnar cells with eosinophilic cytoplasm and minimal nuclear atypia
Given the low incidence, the genetic profile of TLFCK has not been well defined.
The immunohistochemical profile of TLFCK shows positive immunoreactivity for PAX8, EMA, CK7, and vimentin, with a subset of cases reported to express CD10 and PAX2 [71, 76]. The colloid-like material in TLFCK is composed of Tamm-Horsfall glycoprotein , the most abundant protein in normal urine [71, 77]. The main differential diagnosis is metastatic follicular carcinoma of the thyroid; however, TLFCK is negative for thyroid markers TTF-1 and thyroglobulin [6, 71]. The diagnosis of TLFCK is strengthened by the absence of lesions in the thyroid or other locations by imaging [6, 73]. TLFCK should also be distinguished from thyroidization of the kidney, which is characterized by atrophic distal tubules or collecting ducts, with colloid-like hyaline casts that mimic normal thyroid parenchyma. Thyroidization is a benign, widespread, and usually bilateral process that occurs in patients with a history of chronic pyelonephritis, obstructive uropathy, or end-stage renal disease. In contrast, TLFCK presents as a well-circumscribed tumor in a background of unremarkable kidney and is often in patients with no history of renal disease [71].
Succinate Dehydrogenase-Deficient RCC
Introduction
Succinate dehydrogenase (SDH)-deficient renal cell carcinoma is a recently described entity that is not yet widely recognized due to its morphologic overlap with other oncocytic renal neoplasms. Nevertheless, SDH-deficient RCC has been accepted as a provisional entity in the 2013 International Society of Urological Pathology Vancouver Classification of renal tumors, as it represents a distinct renal neoplasm, defined by loss of IHC staining for the B subunit of the SDH mitochondrial complex (the entire complex consists of four subunits: A, B, C, and D) [1, 78, 79]. The tumor is rare, estimated as less than 1 % of all renal epithelial tumors [79], and it occurs in individuals with germ line mutations of the genes that encode the SDH complex [1, 78]. SDH-deficient RCC has been reported more commonly in men, with the mean age of 40 years [78, 79]. Patients with germ line mutation in a SDH subunit gene are also prone to develop paraganglioma/pheochromocytoma, gastrointestinal stromal tumor (GIST), and pituitary adenoma [1, 78–80].
Clinical Presentation
Due to the low incidence of this renal cancer, clinical presentation has yet not been described in detail. Some patients have reported episodic headache and palpitation associated with paraganglioma-related hypertension, with an incidental renal mass discovered on imaging during work-up [81]. Bilateral tumors have been reported [79].
Pathology
On gross examination, this tumor is well circumscribed with a tan to red cut surface [78, 79]. Hemorrhage and microcystic change may be present [78, 79]. Tumor diameter ranges from 2.0 to 20 cm [78]. Both unifocal, multifocal, and bilateral tumors have been described [78, 79].
Microscopically, cells are arranged in solid or nested patterns that contain uniform neoplastic cells with eosinophilic, variably granular cytoplasm [1, 78, 79, 81]. The most distinctive histological feature is the presence of perinuclear cytoplasmic vacuoles and inclusion-like spaces, some of which contain pink flocculent-like material (Fig. 9.6) [78, 79, 81]. Nuclei are typically round with smooth boarders, finely clumped chromatin, and small or inconspicuous nucleoli [78]. Benign tubules or glomeruli are often entrapped at the edges of the tumors [81]. Stromal hemorrhage, fibrosis, and hyalinization are commonly seen [78]. Intratumoral mast cells and aggregates of lymphocytes, as well as necrosis, may be present [78]. Sarcomatoid change has been described [79].
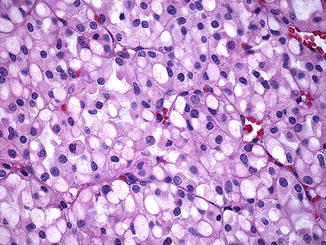
Fig. 9.6




SDHB-associated renal cell carcinoma. Distinctive features include perinuclear cytoplasmic vacuoles and inclusion-like spaces filled with pink secretions
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
