Basic pathology and molecular biology
Neoplasia (the formation and growth of a tumour) may be a
benign or malignant process. Malignant neoplasms, characterized by local invasion of normal tissue or distant spread (metastasis) via lymphatic or vascular channels, may be
primary or secondary. Neoplasms are considered to arise by clonal expansion of a single abnormal cell through uncontrolled aberrant divisions. This cell may be a
stem cell rather than a terminally differentiated cell. Tumour formation results from the loss of balance between cell division and withdrawal from the cell cycle by differentiation or programmed cell death (apoptosis). Signals regulating cell proliferation and interactions come from proteins encoded by messenger
RNA that is in turn transcribed from genomic
DNA. An identifiable precursor lesion may exist.
Urological neoplasms most commonly arise from the lining epithelium of the genitourinary tract. Benign epithelial neoplasms from glandular or transitional epithelium are, respectively, termed
adenoma or
transitional cell papilloma. Malignant epithelial neoplasms are carcinomas; they may be further characterized histologically by prefixing either
adeno if the neoplasm is glandular or
squamous cell or
transitional cell, according to the epithelium from which it has arisen. Carcinomas arise from noninvasive epithelial lesions, some of which are identifiable histologically: in the bladder, it is
flat carcinoma in situ (
CIS) while in the prostate, it is
prostatic intraepithelial neoplasia (PIN). Connective tissue neoplasms are described according to their components, adding benign (-oma) or malignant (-sarcoma) suffixes. For example, a benign neoplasm composed of blood vessels, fat, and smooth muscle is an
angiomyolipoma; a malignant neoplasm composed of smooth muscle is a
leiomyosarcoma. Genitourinary sarcomas are rare, constituting 1% of all neoplasms.
There are exceptions. In the testis, the most common primary neoplasms arise from seminiferous tubules and are termed germ cell tumours. Rarely, primary malignant lymphoma can arise in the testis. In the kidney, the childhood Wilms’ tumour arises from the embryonic mesenchyme of the metanephric blastema while the benign oncocytoma is thought to arise from cells of the collecting ducts.
Secondary malignant neoplasms within urological tissues are uncommon; they may arise by direct invasion from adjacent tissues (for example, adenocarcinoma of the sigmoid colon may invade the bladder) or hae-matogenous metastasis from a distant site such as the lung.
Neoplasia is a genetic disease: it may be hereditary or sporadic, depending on whether the genetic abnormalities are constitutional (germ-line) or somatic (acquired). Hereditary tumours tend to appear at a younger age than their sporadic counterparts and are often multifocal due to an underlying constitutional genetic abnormality.
Genetic and epigenetic abnormalities may promote tumour development or growth in a number of ways.
Activation (overexpression) of
oncogenes encoding transcription factors,
e.g. c-myc.
Inactivation (reduced expression) of
tumour suppressor genes; their diverse protein products stabilize the cell, ensuring differentiation
and a finite lifespan in which it performs its function. Inactivation of such genes by deletion or mutation may result in loss of this negative growth control. For example, PTEN (chromosome 10q) is a prostate tumour suppressor gene, encoding a phosphatase that is active against protein and lipid substrates. It is present in normal epithelium, but is commonly reduced in prostate cancer due to allele loss of chromosome 10q. It inhibits one of the intracellular signalling pathways, PI3 kinase-Akt, that is essential for cell cycle progression and cell survival. Inactivation of PTEN, therefore, promotes cell immortalization and proliferation.
Overexpression of
peptide growth factors, e.g. insulin-like growth factor type 1 in prostate cancer or the highly angiogenic vascular endothelial growth factor in renal cancer.
Promoter methylation or acetylation inactivating genes encoding detoxification enzymes,
e.g GSTP1.
Gene fusions: a translocation occurs during mitosis to bring a promoter gene adjacent to a transcription factor gene on a particular chromosome, resulting in overexpression of this factor and abnormal positive growth control,
e.g. TMPRSS2-ERG fusions are found in 50% of prostate cancers.
MicroRNA: tissue-specific, non-coding, short
ssRNA; regulate gene expression by interacting with
mRNA; multifunctional, measurable, and potentially reversible; postulated to be the key to individualized cancer treatment.
Interest in the molecular pathology of urological neoplasia has begun to result in the development of screening tests for hereditary diseases, diagnostic or prognostic gene profiling, and new strategies for treatment. Examples include the PCA3 test for prostate cancer and the development of targeted therapies for advanced renal cancer.
Wilms’ tumour and neuroblastoma
Wilms’ tumour (nephroblastoma)
First described by the German surgeon, Max Wilms (1867-1918), this is a rare childhood tumour, affecting 1 in 10 000 children. It is, however, the commonest intra-abdominal tumour of childhood (20% of all childhood malignancies) and it represents 80% of all genitourinary tumours affecting children under 15y. Male and female are equally affected, 20% are familial, and 5% are bilateral. Seventy-five percent present under the age of 5y. Children of African descent are at greatest risk.
Pathology and staging
Wilms’ tumour is a soft pale grey tumour (it looks like brain). It contains metanephric blastema, primitive renal tubular epithelium, and connective tissue components. Two distinct histological subtypes are described: favourable (well differentiated) and anaplastic (poorly differentiated).
In at least 20% of cases, mutation or deletion of both copies (alleles) of the chromosome 11p13 WT1 tumour suppressor gene results in tumouri-genesis. The familial disease exhibits autosomal dominant inheritance, but is recessive at the cellular level. Affected family members harbour a germ-line WT1 mutation, conferring susceptibility. One further ‘hit’ is required while two ‘hits’ are required to cause the sporadic disease. This explains why hereditary Wilm’s tumours tend to develop multifocally and at a slightly younger age than its sporadic counterpart. Mutations of three further genes, WT2 (11p15.5), WTX (on the X chromosome), and CTNNB1 account for a further 30% of cases. Loss of chromosome 1p and 16q alleles defines a subgroup with worse prognosis.
Stage I Wilms’ tumour (43% of patients)—at least one of the following criteria must be met.
Tumour is limited to the kidney and is completely excised.
The surface of the renal capsule is intact.
The tumour is not ruptured or biopsied (open or needle) prior to removal.
No involvement of extrarenal or renal sinus lymph-vascular spaces.
No residual tumour apparent beyond the margins of excision.
Metastasis of tumour to lymph nodes not identified.
Stage II Wilms’ tumour (23% of patients)—at least one of the following criteria must be met.
Tumour extends beyond the kidney, but is completely excised.
No residual tumour apparent at or beyond the margins of excision.
Any of the following conditions may also exist.
Tumour involvement of the blood vessels of the renal sinus and/or outside the renal parenchyma.
The tumour has been biopsied prior to removal or there is local spillage of tumor during surgery, confined to the flank.
Extensive tumour involvement of renal sinus soft tissue.
Stage III Wilms’ tumour (23% of patients)—at least one of the following criteria must be met.
Unresectable primary tumour.
Lymph node metastasis.
Tumour is present at surgical margins.
Tumour spillage involving peritoneal surfaces, either before or during surgery, or transected tumour thrombus.
Stage
IV Wilms’ tumour (10% of patients) is defined as the presence of haematogenous metastases (lung, liver, bone, or brain) or lymph node metastases outside the abdominopelvic region.
Stage
V Wilms’ tumour (5% of patients) is defined as bilateral renal involvement at the time of initial diagnosis.
Presentation
Ninety percent have a mass, 33% complain of abdominal or loin pain, 30-50% develop haematuria, 50% are hypertensive. Fifteen percent of patients exhibit other anomalies such as hemihypertrophy/macroglos-sia (Beckwith-Wiedemann syndrome), gonadal dysgenesis/nephropathy (Denys-Drash syndrome) aniridia/retardation (WAGR), and fetal overgrowth (Perlman’s syndrome).
Investigations
The first-line investigation for a child with an abdominal mass or haematuria is ultrasound which will reveal a renal tumour. Further to diagnostic imaging, staging is obtained by
CT, including the chest. Needle biopsy is avoided.
Treatment and prognosis
Children with renal tumours should be managed by a specialist paediatric oncology centre. Staging nephrectomy, with or without preoperative or post-operative chemotherapy, remains the mainstay of treatment. The chemotherapy most frequently used is vincristine and doxorubicin. Flank irradiation may be used in higher stage tumours. Survival is generally good at 92% overall, ranging from 55% to 97%, according to stage at presentation and histology.
Neuroblastoma
The most common extracranial solid tumour of childhood. Eighty percent are diagnosed <4y old. The tumour is of neural crest origin; 50% occur in the adrenal gland and most of the remainder arise along the sympathetic trunks.
Presentation
Systemic symptoms and signs are common: fever, abdominal pain/distension, mass, weight loss, anaemia, and bone pain. Retro-orbital metastases may cause proptosis.
Imaging and staging
Ultrasound initially;
CT of chest and abdomen. Calcification in tumour helps distinguish neuroblastoma from Wilms’ tumour.
MIBG scans are very sensitive for detection of neuroblastomas (
Table 7.1).
Treatment and prognosis
Surgical excision; radiotherapy; combination chemotherapy, possibly with autologous bone marrow transplantation. Stage 4S tumours may resolve with little or no treatment. Prognosis is poor, except for stages 1 and 4S disease.
Radiological assessment of renal masses
Abdominal
USS is the first-line investigation for a patient with loin pain or a suspected renal mass. The size resolution for renal masses is 1.5cm, exhibiting variable echo patterns. Ultrasound may also detect renal cysts, most of which are simple: smooth-walled, round or oval, without internal echoes, and complete transmission with a strong acoustic shadow posteriorly. If the cyst has a solid intracystic element, septations, an irregular or calcified wall, further imaging with
CT is indicated. Ten to twenty-five percent of
RCC contain cysts. Yale radiologist, Morton Bosniak, developed the following radiological classification of renal cysts in
Table 7.2.
1If a renal mass is detected by
USS, a thin slice or helical
CT scan before and after
IV contrast is the most important investigation for characterization and staging. Around 90% of solid-enhancing renal masses will be
RCC. Ten percent of
RCC will contain calcifications or fat. Even relatively avascular renal carcinomas enhance by 10-25
* Hounsfield units. Occasionally, an isodense, but enhancing, area of kidney is demonstrated: this is termed ‘pseudotumour’ and may correspond to a harmless hypertrophied cortical column (of Bertin) or dysmorphic segment.
CT may mislead with respect to liver invasion (rare) due to ‘partial volume effect’; real-time ultrasound is more accurate. Lymphadenopathy >2cm is invariably indicative of metastasis.
MRI with gadolinium contrast may be used for imaging the
IVC, locally advanced disease, renal insufficiency, or for patients allergic to iodinated contrast. Doppler
USS may also evaluate
IVC tumour thrombus.
Renal arteriography is seldom used in the diagnostic setting, but may be helpful to delineate the number and position of renal arteries in preparation for nephron-sparing surgery or surgery for horseshoe kidneys.
Ultrasound or CT-guided fine needle aspiration (FNA) or needle biopsy
This is increasingly indicated due to the trend in managing small masses with surveillance or minimally invasive ablative therapies. Also, a histological diagnosis is usually required prior to treating inoperable patients with systemic therapies. Needle biopsy is highly specific, but less sensitive for detecting malignancy: 80% of biopsy cores are diagnostic, of which 75% are
RCC. Repeat biopsy is diagnostic in 80%. There are also risks of haemorrhage (5%) and tumour spillage (rare).
FNA is useful for aspiration of renal abscess or infected cyst or to diagnose suspected lymphoma or metastatic lesions.
Table 7.3 shows a practical radiological classification of renal masses.
It has been suggested that abdominal
USS could be used as a
screening test for early detection and treatment of
RCC; this has been piloted in Germany and Japan. While there is currently no plan for population screening in the
UK, it would be appropriate to offer
USS to high-risk individuals such as relatives of
VHL syndrome patients.
* Hounsfield units are a measure of X-ray attenuation applied to CT scanning: -1000 units equates with air, 0 units equates with water, and +1000 equates with bone.
1 Bosniak MA (1986) The current radiological approach to renal cysts. Radiology 158: 1-10.
Benign renal masses
The most common (70%) are simple cysts, present in >50% of those aged >50y. Rarely symptomatic, treatment by aspiration or laparoscopic deroofing is seldom considered.
Most benign renal tumours are rare; the two most clinically important are oncocytoma and angiomyolipoma.
Oncocytoma
This is uncommon, accounting for 3-7% of renal tumours. Males are twice as commonly affected as females. They occur simultaneously with
RCC in 7-32% of cases.
Pathology
Oncocytomas are spherical, capsulated, brown/tan colour, mean size 4-6cm. Half contain a central scar. They may be multifocal and bilateral (4-13%) and 10-20% extend into perinephric fat. Histologically, they comprise aggregates of eosinophillic cells thought to arise from intercalated cells of the collecting duct. Cells are packed with mitochondria, mitoses are rare, large nucleoli are present; they are considered benign, not known to metastasize. There is often loss of the Y chromosome.
Presentation
Oncocytomas often (83%) present as an incidental finding or with loin pain or haematuria.
Investigations
Oncocytoma cannot often be distinguished radiologically from
RCC; they may coexist with
RCC. Rarely, they exhibit a ‘spoke-wheel’ pattern on
CT scanning, caused by stellate central scar. Percutaneous biopsy is not usually recommended since there is often continuing uncertainty about the diagnosis. The main
differential diagnosis of renal oncocytoma is
chromophobe RCC oncocytic variant which, like the renal oncocytoma, has eosinophilic cytoplasm, but has perinuclear clearing and typically, some degree of
nuclear atypia.
Angiomyolipoma (AML)
Eighty percent of these benign clonal neoplasms (PEComa, formerly considered as a hamartoma) occur sporadically, mostly in middle-aged females. Twenty percent are in association with tuberous sclerosis (
TS), an autosomal dominant syndrome characterized by mental retardation, epilepsy, adenoma sebaceum, and other hamartomas. Up to 80% of
TS patients develop AMLs, mean age 30y, 66% female, frequently multifocal, and bilateral.
Pathology
AML is composed of perivascular epithelioid cells (
PEC) containing blood vessels, immature smooth muscle, and fat. They are always considered benign although extrarenal AMLs have been reported in venous system, hilar lymph nodes, and liver. Macroscopically, it looks like a well circumscribed lump of fat. Solitary AMLs are more frequently found in the right kidney.
Presentation
AMLs frequently present as incidental findings (>50%) on
USS or
CT scans. They may present with flank pain, palpable mass, or painless haematuria. Massive and life-threatening retroperitoneal bleeding occurs in up to 10% of cases (Wunderlich’s syndrome).
Investigations
Ultrasound reflects from fat, hence a characteristic bright echo pattern. This does not cast an ‘acoustic shadow’ beyond, helping to distinguish an
AML from a calculus.
CT shows fatty tumour as low density (Hounsfield units <10) in 86% of AMLs. If the proportion of fat is low, a definite diagnosis cannot be made as other renal tumours may contain fat. Measurement of the diameter is relevant to treatment.
Renal cell carcinoma: pathology, staging, and prognosis
RCC is adenocarcinoma of the renal cortex, believed to arise from the proximal convoluted tubule (although the majority of
VHL gene deletions occur in the distal tubule). Usually tan-coloured, lobulated, and solid, 7% are multifocal, 1-2% bilateral, 10-20% contain calcification, and 10-25% contain cysts or are predominantly cystic. There may be zones of haemorrhage, necrosis, and scarring. Rarely grossly infiltrative, they are usually circumscribed by a pseudocapsule of compressed tissue.
Spread is by: direct extension to adrenal gland (7.5% in tumours >5cm), through the renal capsule (25%), into renal vein (up to 44%),
IVC (5%), right atrium; by lymphatics to hilar and para-aortic lymph nodes; haematogenously to lung (75%), bone (20%), liver (18%), and brain (8%).
Histological classification of RCC
Conventional (80%): arise from the proximal tubule; highly vascular; clear cells (glycogen, cholesterol) or granular (eosinophillic cytoplasm, mitochondria); involves loss of
VHL, PBRM1, and others genes on chromosome 3.
Papillary (10-15%): papillary, tubular, and solid variants; 40% multifocal; small incidental tumours could equate with Bell’s legendary ‘benign adenoma’; trisomy 7, 16, 17.
Chromophobe (5%): arises from the cortical portion of the collecting duct; possesses a perinuclear halo of microvesicles; hypodiploid with loss of chromosomes 1, 2, 6, 10, 13, 17, 21.
Collecting duct (Bellini): rare, young patients, poor prognosis.
Medullary cell: rare, arises from calyceal epithelium; young sickle cell sufferers; poor prognosis.
‘Sarcomatoid” describes an infiltrative poorly differentiated variant of any type in 5-25%. Coagulative necrosis is seen in 30%. Array-based karyotyping performs well on paraffin-embedded tumours and can be used to identify characteristic chromosomal aberrations in renal tumours with challenging morphology.
Genetic changes associated with
RCC are described on
 p. 250
p. 250.
RCC is an unusually immunogenic tumour, expressing numerous antigens (
e.g. RAGE-1, MN-9). Reports of spontaneous regression, prolonged stabilization, and complete responses to immunotherapy support this. Tumour-infiltrating lymphocytes are readily obtained from RCCs, including T-helper, dendritic, natural killer, and cytotoxic T cells.
RCC is also unusually vascular, overexpressing angiogenic factors, principally
VEGF, but also
bFGF and TGF-β.
Grading is by the Fuhrman system (1 = well differentiated; 2 = moderately differentiated; 3 and 4 = poorly differentiated), based on nuclear size, outline, and nucleoli. It is an independent prognostic factor.
Staging
Staging is by the
TNM classification following histological confirmation of the diagnosis (see
Table 7.4 and
Fig. 7.1). All rely upon physical examination and imaging; the pathological classification (prefixed ‘p’) corresponds to the
TNM categories.
Staging is the most important prognostic indicator for RCC.Factors for
RCC survival include:
Fuhrman grade, necrosis, or sarcomatoid features.
Performance status and systemic symptoms.
Molecular factors (under investigation:
VEGF,
HIF-1, p53, gene expression profiling).
A prognostic nomogram has been developed to predict 5y probability of treatment failure for patients with newly diagnosed
RCC. It is available for download at:
http://www.mskcc.org/mskcc/html/6156.cfm.
Renal cell carcinoma: epidemiology and aetiology
Renal cell carcinoma (
RCC) (also known as hypernephroma since it was erroneously believed to originate in the adrenal gland, clear cell carcinoma, and Grawitz tumour) is the commonest of renal tumours, constituting 2-3% of all cancers. It is an adenocarcinoma, accounting for 85% of renal malignancies; the remainder are
TCC (10%), sarcomas, Wilms’, and other rarities (5%). It occurs in sporadic (common) and hereditary (rare) forms.
Incidence, mortality, and survival
In the
UK, both incidence and mortality are rising, with 8228 patients diagnosed (compared with 3676 patients in 1999) and 3848 deaths in 2008.
RCC is the most lethal of all urological tumours, approximately 50% of patients dying of the condition; it is the tenth most common cause of cancer death. Relative 5y survival, heavily dependent on stage at diagnosis, is 50% while 10y survival fell to 43% for
UK patients. Survival has increased since the 1970’s. As with most cancers, there is a steady fall in survival with advancing age at diagnosis: rates for patients under 50y are twice that for patients over 80.
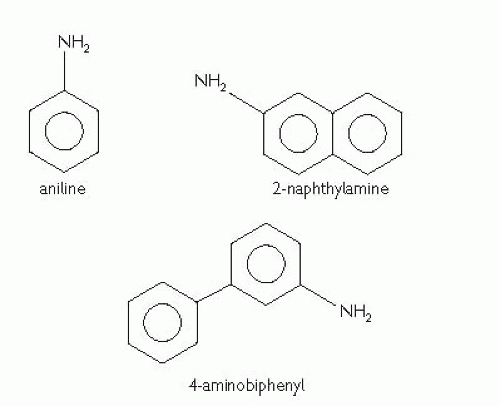
Aetiology
Males are affected 1.5 times as commonly as females; peak incidence of sporadic
RCC is between 60-70y of age.
Environmental
Studies have shown associations with cigarette pipe or cigar
smoking (1.4-2.3-fold risk),
renal failure and dialysis (30-fold risk),
obesity, hypertension (1.4-2-fold risk), urban dwelling, low socio-economic status, tobacco chewing, occupational
asbestos and cadmium exposure, the analgesic
phenacitin, thorium dioxide, and sickle cell trait (medullary carcinoma only).
Nutrition is considered important: Asian migrants to western countries are at increased risk of
RCC; vitamins A, C and E, and fruit/vegetable consumption are protective. Anatomical risk factors include polycystic and horseshoe kidneys.
Genetic
VHL syndrome: 50% of individuals with this autosomal dominant syndrome, characterized by phaeochromocytoma, renal and pancreatic cysts, and cerebellar haemangioblastoma, develop
RCC, often bilateral and multifocal. Patients typically present in 3rd, 4th, or 5th decades.
VHL syndrome occurs due to loss of both copies of a tumour suppressor gene at chromosome 3p25-26; this and other 3p genes (RASSF1A; PBRM1) are implicated in causing >80% of sporadic RCCs. Inactivation of the
VHL gene leads to effects on gene transcription, including dysregulation of hypoxia inducible factors 1 and 2, intracellular proteins that play an important role in the cellular response to hypoxia and starvation. This results in an upregulation of
VEGF, the most prominent angiogenic factor in
RCC, explaining why some RCCs are highly vascular and enabling targeted treatment approaches (see
 p. 258
p. 258).
A
papillary variant of
RCC also has an autosomal dominant familial component, characterized by trisomy 7 and 17, with activation of the c-
MET proto-oncogene. c-
MET is the receptor tyrosone kinase for hepatocyte growth factor which regulates epithelial proliferation and differentiation in a wide variety of organs, including the normal kidney.
Mutations of the
FLCN gene on chromosome 17p results in the autosomal dominant Birt-Hogg-Dubé syndrome. This rare disease is characterized by benign tumours of hair follicles (mainly facial), pulmonary cysts, pneumothoraces, and renal tumours, including oncocytomas and
RCC.
Screening for RCC
Aside from investigating the upper urinary tracts for non-visible asymptomatic haematuria, there is little to support population screening for
RCC using
USS, given that a large study of 10 000 men aged >40y yielded
RCC in only 0.1%.
Renal cell carcinoma: presentation and investigation
At least half of all RCCs are detected incidentally on abdominal imaging carried out to investigate vague or unrelated symptoms. Thus, there has been a downward stage migration at diagnosis since ultrasound and
CT scanning came into routine use in the 1980’s.
Presentation
History: of the symptomatic RCCs diagnosed, 50% of patients present with haematuria, 40% with loin pain, 25% of patients notice a mass, and 30% have symptoms or signs of metastatic disease, including bone pain, night sweats, fatigue, weight loss, and haemoptysis. Less than 10% of patients exhibit the classic triad of haematuria, pain, and abdominal mass. Less common presenting features include pyrexia of unknown origin (9%), acute varicocoele due to obstruction of the testicular vein by tumour within the left renal vein (2-5%), and lower limb oedema due to venous obstruction. Paraneoplastic syndromes due to ectopic hormone secretion by the tumour occur in 30% of patients; these may be associated with any disease stage (
Table 7.6).
Clinical examination: may reveal abdominal mass, cervical lymphadenopathy, non-reducing varicocele, or lower limb oedema (both suggestive of venous involvement).
Investigations
Radiological evaluation: of haematuria, loin pain, and renal mass is described on
 pp. 242
pp. 242 and 270, together with discussion of the role of
needle biopsy.
Urine cytology and culture: should be normal.
FBC: may reveal polycythaemia or anaemia.
Serum creatinine and electrolytes, calcium, and liver function tests: are essential.
When
RCC is diagnosed radiologically, staging
chest CT will follow and
bone scan, if clinically indicated. Any suggestion of renal vein or
IVC involvement on
CT may be further investigated with
Doppler USS or MRI. Angiography may be helpful in planning partial nephrectomy or surgery for horseshoe kidneys. Contralateral kidney function is assessed by the uptake and excretion of
CT contrast and the serum creatinine. If doubt persists,
isotope renography is used.
Renal cell carcinoma (localized): surgical treatment I
Surgery is the mainstay of treatment for
RCC. Increasing diagnosis of smaller, early stage
RCC and the concept of cytoreductive surgery for advanced
RCC has impacted on investigation and surgical treatment strategies while reduction in mortality remains elusive.
Radical nephrectomy
This remains the gold standard treatment of T2-4
RCC and in T1
RCC in patients unsuitable for
PN. There is no difference in outcome favouring a specific surgical approach so the default is now laparoscopic for localized
RCC. In the case of upper pole or T2 tumours, adrenalectomy is also necessary.
Laparoscopic approach: has become a widely available option in centres treating
RCC. Approaches are either transperitoneal or retroperitoneal. The specimen is removed whole or morselated in a bag through an iliac incision. Advantages over open surgery include
less pain, reduced hospital stay, and quicker return to normal activity. Morbidity is reported in 8-38% of cases, including
PE and poorly understood effects on renal function. Long-term (10y) results are equivalent to those obtained by open surgery; cancer-specific survival (
CSS) was 92% in a mixed US series.
Open approach: this should be carried out only for large or locally advanced RCCs. The aim is to remove all tumour with adequate surgical margins by excising the kidney with Gerota’s fascia, vein tumour thrombus, adrenal gland (if invasion indicated by imaging), and limited regional nodes for staging. Surgical approach is transperitoneal (good access to hilar vessels) or thoracoabdominal (for very large or T3c tumours). Following renal mobilization (avoiding tumour manipulation), the ureter is divided; ligation and division of the renal artery or arteries should ideally take place prior to ligation and division of the renal vein to prevent vascular swelling of the kidney. Complications include mortality up to 2% from bleeding or embolism of tumour thrombus; bowel, pancreatic, splenic, or pleural injury.
Post-operative follow-up
aims to detect local or distant recurrence to permit additional treatment, if indicated; incidence is 7% for T1N0M0
RCC, 20% for T2N0M0, and 40% for T3N0M0. After partial nephrectomy, concern will also focus on recurrence in the remnant kidney. There is no consensus regarding the optimal regime, typically stage-dependent 6-monthly clinical assessment and annual
CT imaging of chest and abdomen for 5-10y.
Post-operative prognosis
The Leibovich scoring system groups patients into low, intermediate, or high risk for development of metastasis at 1, 3, 5, 7, and 10y according to tumour stage, size, nuclear grade, presence of necrosis, and regional nodal status. This is particularly useful when selecting patients for trials of adjuvant therapy.
1A nomogram combining prognostic factors for prediction of 5y recurrence risk following surgery can be downloaded at:
http://www.mskcc. org/mskcc/html/6156.cfm
1 Leibovich BC, Blute ML, Cheville JC, et al. (2003) Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: a stratification tool for prospective clinical trials. Cancer 97: 1663-71.
Renal cell carcinoma: surgical treatment II and non-surgical alternatives for localized disease
Localized RCC—lymphadenectomy
Lymph node involvement in
RCC is a poor prognostic factor. Incidence ranges from 6% in T1-2 tumours, 46% in T3A, and 62-66% in higher stage disease. Lymphadectomy at time of nephrectomy may add prognostic information, especially if there is obvious lymphadenopathy, but therapeutic benefit remains unclear. Extended lymphadenectomy adds time and increases blood loss while nodes are clear in about 95% of cases so is not recommended.
Localized RCC: adjuvant therapy
To date, no adjuvant therapy has been shown to improve survival after nephrectomy.
Localized RCC: treatment of local recurrence
Though uncommon, if there is local recurrence in the renal bed after radical nephrectomy, surgical excision remains the preferred treatment choice, provided there are no signs of distant disease. Local recurrence is more common after partial nephrectomy where it can be treated by a further partial or radical nephrectomy.
Localized RCC: alternatives to surgery
Cryosurgery,
HIFU, and Image-guided percutaneous
RFA have the advantage of being outpatient-based, low morbidity, and repeatable; they are currently
recommended only for those patients unfit or unwilling to undergo surgery (since current data show recurrence rates are higher), ideally within clinical trials.
Locally advanced RCC
Disease involving the
IVC, right atrium, liver, bowel, or posterior abdominal wall demand special surgical skills. In appropriate patients, an aggressive surgical approach involving a multidisciplinary surgical team to achieve negative margins appears to provide survival benefit.
Adjuvant treatment: To date no adjuvant therapy has demonstrated any survival benefit, even in those who are predicted to have a higher risk of recurrence. With the advent of the newer tyrosine kinase inhibitors that have demonstrated a benefit in metastatic disease, multiple randomized trials are ongoing and results are awaited.
Metastatic RCC
Nephrectomy has long been indicated for
symptom palliation (pain, haematuria) in patients with metastatic
RCC (if inoperable, arterial embolization can be helpful) and is also performed
prior to systemic therapy, if appropriate. A median survival benefit of 10 months for patients with good performance status treated with cytoreductive nephrectomy prior to immunotherapy (interferon-
α) has been reported. Studies are ongoing to investigate whether there is a similar benefit to cytoreductive nephrectomy with the tyrosine kinase inhibitors. Currently, the standard practice is to recommend it, extrapolating from the cytokine era. Patients should be recruited to the studies that are investigating cytoreductive nephrectomy.
Resection of a solitary metastasis is an appropriate option for a small number of patients, usually a few months after nephrectomy, to ensure the lesion has remained solitary.
1 Jewett MA, Mattar K, Basiuk J, et al. (2011) Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 60: 39-44.
Renal cell carcinoma: management of metastatic disease
Approximately 25% of patients with
RCC have metastatic disease at presentation; a further 30% progress subsequently to this stage following nephrectomy.
Surgery: despite the rare possibility of spontaneous metastatic regression (<5%) following nephrectomy, it was rarely undertaken except to relieve local symptoms of pain or haematuria. The role of nephrectomy in metastatic
RCC is discussed on
 p. 257
p. 257.
Metastasectomy may be of benefit to the 1.5-3% of patients who develop a solitary metastasis (particularly in lung, adrenal, or brain) following nephrectomy.
Angiogenesis (signal transduction) inhibitors
As discussed earlier (see
 p. 250
p. 250), most RCCs are highly angiogenic so fortunately become good therapeutic targets for angiogenesis inhibitors. Via its cell surface receptor (
VEGFR),
VEGF is a pro-angiogenic peptide growth factor that activates the PI3kinase/AKT signal transduction pathway, which is one of three major receptor tyrosine kinase (
RTK) signalling pathways.
VEGF is overexpressed in most sporadic
RCC as a result of
HIF-1 overexpression caused by inactivation of the
VHL tumour suppressor gene. In randomized trials, two well-tolerated oral multi-
RTK inhibitors, sunitinib and pazopanib, have demonstrated significant benefit in the first-line metastatic setting, prolonging progression-free survival (
PFS) in metastatic
RCC patients by 3-8 months compared with interferon alpha (
IFNα) or placebo. The
UK NICE approved both in 2009 and 2011, respectively. Complete responses are rare, partial responses modest (30-40%), they also stabilize the disease in approximately 30% of patients.
1 Pazopanib is effective as second-line treatment (prior cytokine therapy).
A further randomized trial demonstrated >3-month survival advantage of temsirolimus, an inhibitor of cytoplasmic mTOR kinase (a downstream component of the same pathway) in metastatic
RCC patients compared with
IFNα.
2 This is currently recommended for first-line treatment of poor risk disease. For second-line, everolimus is an orally available mTOR inhibitor: it confers a 2-month
PFS over placebo when used for patients failing the treatments. However,
NICE has not approved its use (2011).
VEGF antibodies
Bevacizumab is a humanized monoclonal antibody that binds to
VEGFR. A phase III randomized trial demonstrated a median 31% response with bevacizumab +
IFNα compared with
IFNα alone, with a 4.8-month PDS
advantage for low and intermediate risk patients. This combination is an option for first-line treatment.
These agents represent a major advance in the first- and second-line treatment of metastatic
RCC.
There are multiple newer thymidine kinase inhibitors (TKI) that are also currently being investigated.
Immunotherapy
The immunogenicity of
RCC is discussed on
 p. 246
p. 246. The first cytokines to be used therapeutically to activate anti-tumour immune response were interferons and subsequently
IL-2. Randomized studies in the 1990’s demonstrated modest response rates (10-20%) after
systemic immunotherapy using these cytokines alone and in combination; toxicity could be severe. Responses were more likely in patients with good performance status, prior nephrectomy, and small-volume metastatic burden. An MRC trial of
IFNα vs medroxyprogesterone demonstrated a 2.5-month survival advantage in the immunotherapy group. The use of immunotherapy has been overshadowed recently by the development of
RTK inhibitors, although there may still be a role for
IL-2 in a very select group of patients and is still being used for appropriate patients (excellent performance status, small volume lung only metastases, and no prior treatment).
Chemotherapy: little role in
RCC; ineffective due to high multidrug resistance P glycoprotein expression.
Radiotherapy: useful for palliation of metastatic lesions in bone and brain and in combination with surgery for spinal cord compression.
Palliative care
Steroids (
e.g. dexamethazone 4mg
qds) improve appetite and mental state, but are unlikely to impact on tumour growth. The involvement of multidisciplinary uro-oncology, palliative, and primary care teams is essential to support these patients and their relatives.
1 Motzer RJ, Hutson TE, Tomczak P, et al. (2007) Sunitimib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 356: 115-24.
2 Sternberg CN, Davis ID, Mardiak J, et al. (2010) Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 28: 1061-8.
3 Hudes G, Carducci M, Tomczak P, et al. (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356: 2271-81.
Upper urinary tract transitional cell carcinoma (UUT-TCC)
UUT-TCC accounts for 90% of upper urinary tract tumours, the remainder being benign inverted papilloma, fibroepithelial polyp, squamous cell carcinoma (associated with longstanding staghorn calculus disease), adenocarcimona (rare), and various rare non-urothelial tumours, including sarcoma.
TCC of the renal pelvis is uncommon, accounting for 10% of renal tumours and 5% of all
TCC. Ureteric
TCC is rare, accounting for only 1% of all newly presenting
TCC. Half are multifocal; 75% located distally while only 3% are located in the proximal ureter.
Risk factors are similar to those of bladder
TCC (see
 p. 264
p. 264).
Males are affected three times as commonly as females.
Incidence increases with age.
Smoking confers a 2-fold risk and there are various occupational causes.
Phenacetin ingestion.
There is a high incidence of
UUT-TCC in families from some villages in Balkan countries
(‘Balkan nephropathy’) that remains unexplained.
Lynch syndrome (hereditary non-polyposis colon cancer) is an autosomal dominant condition caused by a
DNA mismatch repair defect; it is associated with various cancers, including
UUT-TCC, most in middle-aged females.
Pathology and grading
The tumour usually has a papillary structure, but occasionally solid. It is bilateral in 2-4%. It arises within the renal pelvis, less frequently in one of the calyces or ureter. Histologically, features of
TCC are present; grading is as for bladder
TCC. Spread is by direct extension, including into the renal vein and vena cava; lymphatic spread to para-aortic, para-caval, and pelvic nodes; bloodborne spread, most commonly to liver, lung, and bone.
Presentation
Painless total haematuria (80%).
Loin pain (30%), often caused by clots passing down the ureter (‘clot colic’).
Asymptomatic when detected, associated with synchronous bladder
TCC (4%).
At follow-up, approximately 50% of patients will develop a metachronous bladder
TCC and 2% will develop contralateral upper tract
TCC.
Investigations
Ultrasound is excellent for detecting the more common renal parenchymal tumours, but not sensitive in detecting tumours of the renal pelvis or ureter.
Diagnosis is usually made on
urine cytology and
CTU, respectively, revealing malignant cells and a filling defect in the renal pelvis or ureter. If doubt exists, selective ureteric urine cytology,
retrograde ureteropyelgraphy, or
flexible
ureterorenoscopy with
biopsy are indicated. Some surgeons prefer to have histological proof of malignancy prior to treatment. Additional staging is obtained by chest
CT and occasionally, isotope bone scan.
Staging uses the
TNM (2009) classification (
Table 7.7) following histological confirmation of the diagnosis. All rely on physical examination and imaging, the pathological classification corresponding to the
TNM categories.
Metastatic disease
Systemic combination chemotherapy (platinum-based): for unresectable or metastatic disease is associated with a 30% total or partial response at the expense of moderate toxicity.
Palliative surgery or arterial embolization: may be necessary for troublesome haematuria. Radiotherapy is generally ineffective.
Muscle-invasive
UUT-TCC, constituting 60% of new presentations, have a poor prognosis. The following are recognized prognostic factors, in descending order of importance:
Bladder cancer: pathology, grading, and staging
Benign tumours of the bladder, including inverted urothelial papilloma and nephrogenic adenoma, are uncommon.
The vast majority of primary bladder cancers are malignant and epithelial in origin.
1-7% are
SCC; 75% are
SCC in areas where schistosomiasis is endemic.
2% are adenocarcinoma.
Small cell and spindle cell carcinomas (rare).
Other rare primary tumours include phaeochromocytoma, melanoma, lymphoma, and sarcoma arising within the bladder muscle.
Secondary bladder cancers are mostly directly spread by adenocarcinomas from the gut, prostate, kidney or ovary, or squamous carcinoma of the urerine cervix.
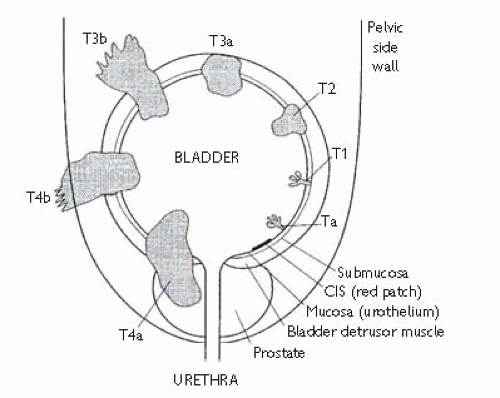
Tumour spread is:
Direct: tumour growth to involve the detrusor, the ureteric orifices, prostate, urethra, uterus, vagina, perivesical fat, bowel, or pelvic side walls.
Implantation: into wounds/percutaneous catheter tracts.
Lymphatic: infiltration of the iliac and para-aortic nodes.
Haematogenous: most commonly to liver (38%), lung (36%), adrenal gland (21%), and bone (27%). Any other organ may be involved.
Histological grading has traditionally (1973
WHO Classification) been divided into: benign urothelial papilloma; well, moderately, and poorly differentiated (G1, G2, and G3, respectively) carcinoma. Most retrospective studies, clinical trials, and guidelines are based on this classification. The 2004
WHO grading uses cytological/architectural criteria to distinguish flat lesions (hyperplasia, dysplasia, carcinoma
in situ) and raised lesions (urothelial papilloma, papillary urothelial neoplasms of low malignant potential (
PUNLMP), low-grade and high-grade urothelial carcinomas). The 2004 system is more reproducible, but is as yet not proven to be of better prognostic value than the 1973 system. Hence, both systems are used in contemporary clinical practice, with G2 tumours being called either low-grade or high-grade.
Staging is by the
TNM (2009) classification (
Table 7.9 and
Fig. 7.3). All rely upon physical examination and imaging, the pathological classification (prefixed ‘p’) corresponding to the
TNM categories.
TCC: may be single or multifocal. Because 5% of patients will have a synchronous upper tract
TCC and metachronous recurrences may develop after several years, the urothelial ‘field change’ theory of polyclonality has been favoured over the theory of tumour monoclonality with transcoelomic implantation (seeding).
Primary
TCC is either
non-muscle invasive (formerly known as ‘superficial’) or
muscle-invasive.
70% of tumours are
papillary, usually G1 or G2, exhibiting at least seven transitional cell layers covering a fibrovascular core (normal transitional epithelium has approximately five cell layers).
Papillary TCC is usually
superficial, confined to the bladder mucosa (Ta) or submucosa (T1); 10% of patients subsequently develop muscle-invasive or metastatic disease. However, G3T1 tumours are more aggressive, with 40% subsequently upstaging.
10% of
TCC have
mixed papillary and solid morphology and 10% are
solid. These are usually G3, half of which are
muscle-invasive at presentation.
10% of
TCC is flat
CIS. This is poorly differentiated carcinoma, but confined to the epithelium and associated with an intact basement membrane; 50% of
CIS lesions occur in isolation; the remainder occurs in association with muscle-invasive
TCC.
CIS usually appears as a flat red velvety patch on the bladder mucosa; 15-40% of such lesions are
CIS, the remainder being focal cystitis of varying aetiology. The cells are poorly cohesive, up to 100% of patients with
CIS exhibiting positive urine cytology in contrast to much lower yields (17-72%) with G1/2 papillary
TCC; 40-83% of untreated
CIS lesions will progress to muscle-invasive
TCC, making
CIS the most aggressive form of superficial
TCC.
5% of patients with G1/2
TCC and at least 20% with G3
TCC (including
CIS) have vascular or lymphatic spread.
Metastatic lymph node disease is found in: 0% Tis, 6% Ta, 10% T1, 18% T2 and T3a, 25-33% T3b and T4
TCC.
SCC: is usually solid or ulcerative and muscle-invasive at presentation.
SCC accounts for only 1% of
UK bladder cancers.
SCC in the bladder is associated with chronic inflammation and urothelial squamous metaplasia rather than
CIS. In Egypt, 80% of
SCC is induced by the ova of
Schistosoma haematobium. Five percent of paraplegics with long-term catheters develop
SCC. Smoking is also a risk factor for
SCC. The prognosis is better for bilharzial
SCC than for non-bilharzial disease, probably because it tends to be lower-grade and metastases are less common in these patients.
Adenocarcinoma: is rare, usually solid/ulcerative, G3, and carry a poor prognosis. One-third originate in the urachus, the remnant of the allantois, located deep to the bladder mucosa in the dome of the bladder. Adenocarcinoma is a long-term (10-20+ year) complication of bladder exstrophy and bowel implantation into the urinary tract, particularly bladder substitutions and ileal conduits after cystectomy. There is association with cystitis glandularis rather than
CIS. Secondary adenocarcinoma of the bladder may arise.
Bladder cancer (muscle-invasive): staging and surgical management of localized (pT2/3a) disease
This is a dangerous disease; the untreated 5y survival is just 3%. Management of patients with invasive bladder cancer requires a multidisciplinary team approach, involving case-by-case discussion between the urological surgeon, radiotherapist, and medical oncologist, with support from the pathologist, radiologist, and cancer specialist nurse.
Staging investigations
In the absence of prospective randomized trials comparing the surgical and non-surgical treatments, the options for a patient with newly diagnosed confined muscle-invasive bladder cancer are:
Bladder preserving.
Radical cystectomy with:
Ileal conduit urinary diversion.
Ureterosigmoidostomy urinary diversion.
Continent urinary diversion.
± Neoadjuvant chemotherapy: some evidence of benefit (see
 p. 286
p. 286).
± Neoadjuvant
RT: no evidence of benefit (see
 p. 288
p. 288).
Partial cystectomy is a good option for well selected patients with small solitary disease located near the dome and for urachal carcinoma. Morbidity is less than with radical cystectomy and diversion is not required. The surgical specimen should be covered with perivesical fat, with a 1.5cm margin of macroscopically normal bladder around the tumour. There should be no biopsy evidence of
CIS elsewhere in the bladder.
The bladder must be closed without tension and catheterized for 7-10 days to allow healing. Subsequent review cystoscopies are mandatory to ensure no tumour recurrence.
Radical cystectomy with urinary diversion
This is the most effective primary treatment for muscle-invasive
TCC,
SCC, and adenocarcinoma and can be performed as salvage treatment if
RT has failed. It is also a treatment for G3T1
TCC and
CIS, refractory to
BCG. Any ureteric obstruction caused by the primary tumour will be relieved by the concomitant urinary diversion. However, this is a major undertaking for the patient and surgeon, requiring support from the cancer specialist nurse, stomatherapist, or continence advisor. Preoperative bowel preparation is discouraged by ‘enhanced recovery’ specialists since it is considered to cause unnecessary dehydration without any evidence of benefit.
The procedure: through a midline transperitoneal or extraperitoneal approach, a bilateral pelvic lymphadenectomy is undertaken. The extent of lymphadenectomy ranges from limiting dissection to the obturator fossa to extending the dissection up to the aortic bifurcation. Studies have suggested a survival advantage to the extended approach, provided the primary cancer is stage T2 or less. The findings of frozen section histology may influence the decision to proceed in some cases. The entire bladder is then excised along with perivesical fat, vascular pedicles, and urachus, plus the prostate/seminal vesicles or anterior vaginal wall. The anterior urethra is not excised unless there is prior biopsy evidence of tumour at the female bladder neck or prostatic urethra (when recurrence occurs in 37%). The ureters are divided close to the bladder, ensuring their disease-free status by frozen section histology, if necessary, and anastomosed into the chosen urinary diversion (see
 p. 290
p. 290).
Some centres are pioneering laparoscopic and robot-assisted cystectomy. Advantages include less blood loss, less post-operative analgesia requirement, and reduced hospital stay. However, long operating times, high cost, and technical considerations may limit widespread adoption of this approach. Oncological outcomes are still under evaluation.
Major complications affect 25% of cystectomy patients. These include perioperative death (1.2%), reoperation (10%), bleeding, thromboembolism, sepsis, wound infection/dehiscence (10%), intestinal obstruction or prolonged ileus (10%), cardiopulmonary morbidity, and rectal injury (4%). Erectile dysfunction is likely after cystectomy due to cavernosal nerve injury.
The complications of urinary diversion are discussed on
 p. 290
p. 290.
Post-operative care
Many patients will spend the first 24h in the high dependency unit or
ITU.
Daily clinical evaluations, including inspection of the wound (and stoma, if present), fluid balance, urine and drain outputs, blood count, creatinine/electrolytes, and albumin.
Broad-spectrum antimicrobial prophylaxis.
Venous thromboembolism prophylaxis with TED stockings, pneumatic calf compression, and subcutaneous
LMWH (unfractionated heparin for patients with renal impairment);
NICE (2010) recommends continuing heparin for 28 post-operative days.
Early mobilization within 24h, if possible.
Chest physiotherapy and adequate analgesia is especially important in smokers and patients with chest comorbidity.
Oral intake is commenced early, an integral part of the Enhanced Recovery concept; however, patients may require parenteral nutrition in the presence of
GI complications or prolonged ileus.
Drains are usually sited in the pelvis and near the ureterodiversion anastamosis; ureteric catheters pass from the renal pelves through the diversion and exit percutaneously; a catheter drains the diversion (except in the case of ileal conduit), exiting urethrally or suprapubically.
Most patients stay in hospital for 10-14 days.
Salvage radical cystectomy is technically a more difficult and slightly more morbid procedure. Relatively few patients who have failed primary
RT are suitable for this second chance of a cure; these are fit patients with mobile clinically localized disease.
Efficacy of radical cystectomy
Failure to cure may result from inadequate excision of the primary tumour or presence of metastases (
Table 7.11). Treatment delay should be avoided if possible; cystectomy performed within 3 months of diagnosis (T2
TCC) results in significantly improved survival compared with >3 months.
1 Pathological upstaging of the primary can occur in up to 40% of cases. Lymph node metastases occur in 10% of T1 and up to 33% of T3-4 cancers. The use of neoadjuvant chemotherapy in muscle-invasive disease is discussed on
 p. 288
p. 288.
1 Lee CT, Madii R, Daignault S, et al. (2006) Cystectomy delay more than 3 months from initial bladder cancer diagnosis results in decreased disease specific and overall survival. J Urol 175: 1262-7.
Bladder cancer: urinary diversion after cystectomy
The choice of urinary diversion requires consideration of both
clinical and quality of life (QoL) issues. Patients planned for cystectomy should be informed of the possible options. Contraindications to the continent reconstructive procedures include debilitating neurological and psychiatric illness, short life expectancy, and impaired renal or liver function. These patients must be motivated and able to perform
ISC. Contraindications to orthotopic neobladder include tumour in the prostatic urethra, widespread
CIS, and urethral stricture disease.
The majority of patients report good overall
QoL following urinary diversion. The reconstructive procedures were expected to be better for social functioning compared to the ileal conduit; most
QoL studies have not shown significant differences, although patients with continent diversions generally score more favourably in terms of body image, social activity, and physical function.
Ureterosigmoidostomy
Dating back to 1821, the oldest form of urinary diversion, whereby the ureters drain into the sigmoid colon, either in its native form or following detubularization and reconstruction into a pouch (Mainz II). This diversion requires no appliance (stoma bag, catheter) so remains popular in developing countries. In recreating a ‘cloaca’, the patient may be prone to upper
UTI with the risk of long-term renal deterioration, metabolic hyperchloraemic acidosis, and loose frequent stools. The low-pressure and capacious Mainz II pouch reduces, but does not abolish, these complications.
Ileal conduit
This was developed during the 1940’s by Eugene Bricker of St. Louis; it remains the most popular form of urinary diversion in the
UK. Fifteen
cm of subterminal ileum is isolated on its mesentery and the ureters are anastomosed to the proximal end. The distal end is brought out in the right iliac fossa as a stoma. The native ileum is anastomosed to gain enteral continuity.
Complications of ileal conduit are:
Prolonged ileus.
Urinary leak.
Enteral leak.
Pyelonephriris.
Uretero-ileal stricture.
Stoma problems (20%—skin irritation, stenosis, and parastomal hernia).
Upper tract dilatation (30%).
Patients require stomatherapy support and some find difficulty in adjusting their lifestyle to cope with a stoma bag. Metabolic complications are uncommon.
In post-
RT salvage patients, a jejunal or colonic conduit is used because of concerns about the healing of radiation-damaged ileum. The conduit
may be brought out in the upper abdomen and patients require careful electrolyte monitoring due to sodium loss and hyperkalaemia.
Continent diversion
The advantage is the absence of an external collection device. There are two types of continent diversion.
A continent pouch is fashioned from 60cm of detubularized ileum or right hemicolon. The ureters drain into this low-pressure balloon-shaped reservoir, usually through an antireflux submucosal tunnel. This is drained by the patient via a continent catheterizable stoma, such as the appendix or uterine tube (the Mitrofanoff principle) brought out in the right iliac fossa.
A similarly constructed pouch may be anastomosed to the patient’s urethra to act as an
orthotopic neobladder so that natural voiding can be established and no stoma is necessary. Patients void by relaxing their external sphincter and performing a Valsalva. This neobladder should require no catheter, unless the pouch is too large and fails to empty adequately. In this case, the patient must be prepared to perform
ISC.
Popular ileal pouches include those of Studer (see
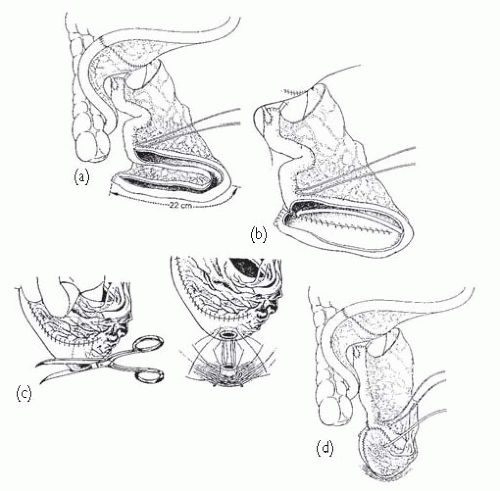
Fig. 7.4), Camey II, and Kock. Ileocaecal pouches include the Indiana and Mainz I. Which one is chosen often comes down to the surgeon’s preference; they carry similar complication risks. Previously irradiated bowel can safely be used to form pouches though complications are more likely.
Complications relating to pouches and neobladders are divided into early (12%) and late (37%). They include:
Urinary leakage and peritonitis.
Pelvic abscess.
Stone formation.
Catheterizing difficulties and stomal stenosis.
Urinary incontinence and nocturnal enuresis (particularly with neobladders).
Pouch ureteric reflux and
UTI.
Ureteropouch anastomotic stricture.
Late neobladder rupture.
Metabolic abnormalities include early fluid and electrolyte imbalances; later, urinary electrolyte absorption may cause hyperchloraemic acidosis and loss of small bowel may result in vitamin B12 deficiency. Metabolic acidosis is less likely in patients with normal renal function; treatment is with sodium bicarbonate and potassium citrate. Annual B12 monitoring should be undertaken with supplementation if necessary.
Adenocarcinoma may develop (5%) in intestinal conduit, neobladder, or sigmoid colon mucosa in the long term due to the carcinogenic bacterial metabolism of urinary nitrosamines. This tends to occur near to the inflow of urine. It is, therefore, advisable to perform annual visual surveillance of urinary diversions after 10y. If the urethra is in situ, annual urethroscopy and cytology is important.
1 Studer UE, Danuser H, Hochreiter W, et al. (1996) Summary of 10 years experience with an ileal low-pressure substitute combined with an afferent tubular isoperistaltic segment. World J Urol 14: 29-39.

 p. 272).
p. 272). p. 274. As a primary treatment, a visually complete tumour resection is adequate for 70% of newly presenting patients with Ta/T1 superficial disease. The remaining 30% of patients experience early recurrence, 15% with upstaging. Because of this, it is standard care that all new patients receive adjuvant treatment with a single dose of post-operative intravesical chemotherapy (usually mitomycin; see
p. 274. As a primary treatment, a visually complete tumour resection is adequate for 70% of newly presenting patients with Ta/T1 superficial disease. The remaining 30% of patients experience early recurrence, 15% with upstaging. Because of this, it is standard care that all new patients receive adjuvant treatment with a single dose of post-operative intravesical chemotherapy (usually mitomycin; see  p. 280). Complications of TURBT are uncommon, including bleeding, sepsis, bladder perforation, incomplete resection, and urethral stricture.
p. 280). Complications of TURBT are uncommon, including bleeding, sepsis, bladder perforation, incomplete resection, and urethral stricture.
 p. 280).
p. 280). http://www.eortc.be/tools/bladdercalculator/.
http://www.eortc.be/tools/bladdercalculator/.
 p. 288).
p. 288). p. 284). Otherwise, cytotoxic chemotherapy (see
p. 284). Otherwise, cytotoxic chemotherapy (see  p. 289) and palliative measures may be considered.
p. 289) and palliative measures may be considered. pp. 363 and 546).
pp. 363 and 546).