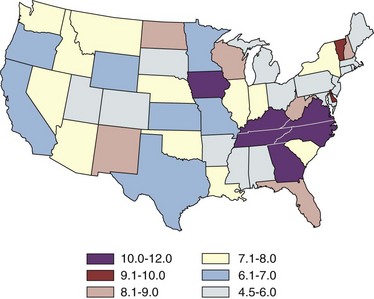
Margaret S. Pearle, MD, PhD, Yair Lotan, MD Although stone disease is one of the most common afflictions of modern society, it has been described since antiquity. With Westernization of global culture, however, the site of stone formation has migrated from the lower to the upper urinary tract and the disease once limited to men is increasingly gender blind. Revolutionary advances in the minimally invasive and noninvasive management of stone disease over the past 2 decades have greatly facilitated the ease with which stones are removed. However, surgical treatments, although they remove the offending stone, do little to alter the course of the disease. Indeed the overall estimated annual expenditure for individuals with insurance claims corresponding to a diagnosis of nephrolithiasis was nearly $2.1 billion in 2000, reflecting a 50% increase since 1994 (Pearle et al, 2005). Given the frequency with which stones recur, the development of a medical prophylactic program to prevent stone recurrences is desirable. To this end, a thorough understanding of the etiology, epidemiology, and pathogenesis of urinary tract stone disease is necessary. The lifetime prevalence of kidney stone disease is estimated at 1% to 15%, with the probability of having a stone varying according to age, gender, race, and geographic location. In the United States, the prevalence of stone disease has been estimated at 10% to 15% (Norlin et al, 1976; Sierakowski et al, 1978; Johnson et al, 1979). Using data derived from the U.S. National Health and Nutrition Examination Survey dataset (NHANES II and III), Stamatelou and colleagues (2003) established a 5.2% prevalence of kidney stone disease from 1988 to 1994, which represents a 37% increase from 1976 to 1980, for which a 3.8% prevalence rate was determined. The finding of an increase in the prevalence of stone disease has been observed by others (Norlin et al, 1976; Yoshida and Okada, 1990; Serio et al, 1999; Trinchieri et al, 2000). In a unique dataset derived from a series of nationwide surveys conducted by the Japanese Society on Urolithiasis Research in cooperation with the Japanese Urological Association, the incidence of first-time stone episodes in the Japanese population has been determined over a 40-year time period (Yasui et al, 2008). The fifth such survey, reported in 2008, estimated the annual incidence of first-time stone events in 2005 and determined trends in stone prevalence using the previous surveys from 1965, 1975, 1985, and 1995. The authors found an increase in the age-adjusted annual incidence of first-time stone events, from 54.2 per 100,000 in 1965 to 114.3 per 100,000 in 2005. Although the incidence increased in all age groups and in both men and women, the age of peak incidence shifted in men from 20 to 49 years in 1965 to 30 to 69 years in 2005 and in women from 20 to 29 years in 1965 to 50 to 79 years in 2005. Stone disease typically affects adult men more commonly than adult women. By a variety of indicators including inpatient admissions, outpatient office visits, and emergency department visits, men are affected two to three times more often than women (Hiatt et al, 1982; Soucie et al, 1994; Pearle et al, 2005). However, there is some evidence that the difference in incidence between men and women is narrowing. Using the National Inpatient Sample dataset representing hospital discharges, Scales and colleagues (2005) found that although overall population-adjusted discharges for a diagnosis of renal or ureteral calculus increased by only 1.6% from 1997 to 2002, discharges for women increased by 17%, whereas discharges for men decreased by 8.1%. This trend reflects a change in the ratio of male-to-female discharges from 1.7 in 1997 to 1.3 in 2002. Indeed, in 2002 hospital discharges were the same for men and women in this dataset. Whether trends in hospital discharges accurately reflect trends in the overall prevalence of the disease, however, is not clear. Stamatelou and colleagues (2003) also noted a slight decrease in the male-to-female ratio of stone disease, from 1.75 (between 1976 and 1980) to 1.54 (between 1988 and 1994) using the NHANES dataset. Racial/ethnic differences in the incidence of stone disease have been observed. Among U.S. men, Soucie and colleagues (1994) found the highest prevalence of stone disease in whites, followed by Hispanics, Asians, and African-Americans, who had prevalences of 70%, 63%, and 44% of whites, respectively. Among U.S. women, the prevalence was highest among whites but lowest among Asian women (about half that of whites). Others found an even higher differential (threefold to fourfold) between whites and African-Americans (Sarmina et al, 1987). Mente and colleagues (2007) attempted to identify genetic influences on stone disease by comparing stone prevalence among different ethnic groups residing in the same geographic region. Using Europeans (Caucasians) as the reference group, the relative risk of calcium stones was higher in individuals of Arabic (OR 3.8, 95% CI 2.7 to 5.2); West Indian (OR 2.5, 95% CI 1.8 to 3.4); West Asian (OR 2.4, 95% CI 1.7 to 3.4); and Latin American (OR 1.7, 95% CI 1.2 to 2.4) origin and significantly lower in those of East Asian (OR 0.4, 95% CI 0.3 to 0.5) and African (OR 0.7, 95% CI 0.5 to 0.9) descent. Interestingly, despite differences in prevalence of stone disease according to ethnicity, Maloney and colleagues (2005) observed a remarkably similar incidence of metabolic abnormalities between white and nonwhite stone formers from the same geographic region, although the distribution of abnormalities differed, suggesting that dietary and other environmental factors may outweigh the contribution of ethnicity in determining stone risk. The gender distribution of stone disease varies according to race. Sarmina and colleagues (1987) noted a male-to-female ratio among whites of 2.3 and among African-Americans of 0.65. Michaels and colleagues (1994) also noted a reversal of the male predisposition to stone disease in Hispanics and African-Americans, reporting a male-to-female ratio of 1.8 among Asians, 1.6 among whites, 0.7 among Hispanics, and 0.5 among African-Americans, among a group of patients undergoing extracorporal shock wave lithotripsy. Soucie and associates (1994) observed a similar trend in the male-to-female ratio of the lifetime incidence of stone disease of 3.4 among Asians, 2.6 among whites, 2.1 among Hispanics, and 1.8 among African-Americans, although the actual ratios for each racial group differed in the two studies. Dall’era and colleagues (2005) reviewed emergency department records to identify patients presenting with symptomatic renal or ureteral calculi and found a male-to-female ratio of 1.17 among Hispanic patients compared with 2.05 for white patients. Stone occurrence is relatively uncommon before age 20 but peaks in incidence in the fourth to sixth decades of life (Marshall et al, 1975; Johnson et al, 1979; Hiatt et al, 1982). It has been observed that women show a bimodal distribution of stone disease, demonstrating a second peak in incidence in the sixth decade of life, corresponding to the onset of menopause (Marshall et al, 1975; Johnson et al, 1979). This finding and the lower incidence of stone disease in women compared with men have been attributed to the protective effect of estrogen against stone formation in premenopausal women, owing to enhanced renal calcium absorption and reduced bone resorption (McKane et al, 1995; Nordin et al, 1999). Indeed, Heller and colleagues (2002) identified lower urinary saturation of calcium oxalate and brushite in women compared with men. Moreover, urinary calcium was lower in women than in men until age 50, after which it reached equivalence in the two groups. Estrogen-treated postmenopausal women had lower urinary calcium and saturation of calcium oxalate than untreated women. However, others have observed a decreasing incidence of stone disease in women from a peak in the late 20s until age 50, after which it remained constant (Hiatt et al, 1982; Curhan et al, 1993). Yasui and colleagues (2008) observed a change in the peak incidence of stone disease in Japanese women from ages 20 to 29 in 1965 to ages 50 to 79 in 2005. Alternatively, Fan and colleagues (1999) found that androgens increased and estrogens decreased urinary and serum oxalate in an experimental rat model, perhaps accounting for the reduced risk of stone formation in women. However, van Aswegen and colleagues (1989) found lower levels of urinary testosterone in stone formers compared with non–stone-forming control subjects, further confusing the issue. The geographic distribution of stone disease tends to roughly follow environmental risk factors; a higher prevalence of stone disease is found in hot, arid, or dry climates such as the mountains, desert, or tropical areas. However, genetic factors and dietary influences may outweigh the effects of geography. Finlayson (1974) reviewed several worldwide geographic surveys and found that areas of high stone prevalence included the United States, British Isles, Scandinavian and Mediterranean countries, northern India and Pakistan, northern Australia, Central Europe, portions of the Malay peninsula, and China. Within the United States, Mandel and Mandel (1989a, 1989b) identified the highest rates of hospital discharge for patients with calcium oxalate stones in the Southeast and for uric acid stones in the East, among the veteran patient population. Soucie and colleagues (1994) found increasing age-adjusted prevalence rates in both men and women going from north to south and west to east, with the highest prevalence observed in the Southeast (Fig. 45–1). After controlling for other risk factors, the authors determined that ambient temperature and sunlight were independently associated with stone prevalence (Soucie et al, 1996). (From Mandel NS, Mandel GS. Urinary tract stone disease in the United States veteran population: II. Geographical analysis of variations in composition. J Urol 1989;142:1516.) Seasonal variation in stone disease is likely related to temperature by way of fluid losses from perspiration and perhaps by sunlight-induced increases in vitamin D. Prince and Scardino (1960) noted the highest incidence of stone disease in the summer months, July through September, with the peak occurring within 1 to 2 months of maximal mean temperatures (Prince et al, 1956). Likewise, Bateson (1973) reported a peak incidence of stone disease between December and March in Australia, corresponding to the summer season. Using data obtained from the Taiwan National Health Insurance Research Database (1999-2003), Chen and colleagues analyzed monthly inpatient and outpatient medical benefit claims for a primary diagnosis of renal or ureteral calculi or renal colic and found that the peak incidence of stone-related claims occurred in July through September, with a sharp decline in claims in October (Chen et al, 2008). Ambient temperature, atmospheric pressure, and hours of sunshine all correlated with monthly stone-related claims, but after adjusting for seasonality, month, and trend, ambient temperature was found to be the most important determinant of stone-related events. The study of military personnel translocated to desert locations has provided a unique opportunity to study the effect of climate on a defined population. Pierce and Bloom (1945) reported that American soldiers in an undisclosed desert location had an increase in symptomatic episodes of renal colic during the summer season. Another study of military personnel who developed symptomatic stones after arrival in Kuwait and Iraq disclosed a mean time interval to stone formation of 93 days (Evan et al, 2005). Finally, Parry and Lister (1975) measured urinary calcium and magnesium levels in soldiers before and 10 days after transfer to the Persian Gulf and noted increased urinary calcium levels from baseline in those soldiers transferred during the summer months but not among those transferred during the “cold season,” which was attributed to sunlight-induced increased production of 1,25-dihydroxyvitamin D3 (1,25[OH]2D3). Thus it is likely that climate and geography influence the prevalence of stone disease indirectly, through effects on temperature and possibly sunlight. Brikowski and colleagues (2008) constructed two alternate models describing the temperature dependence of stone disease on the basis of reported regional stone prevalence rates and corresponding mean annual temperatures (MATs) in order to predict the anticipated change in stone prevalence due to global warming. Prevalence rates obtained from the Second Cancer Prevention Survey of 1982 (Soucie and Coates, 1996) were consistent with a nonlinear, or peaked, relationship between temperature and stone prevalence, while a dataset from the Veteran’s Administration that was analyzed by the Urologic Diseases in America Project more closely approximated a linear fit (Pearle et al, 2005). Using a moderate-severity warming model to predict temperature change due to global warming in the United States, the authors estimated an increase of 1 to 1.5 million lifetime cases of climate-related nephrolithoiasis by 2050. According to the linear model of temperature dependence, the net effect of warming will be a northward expansion of the current-day “stone belt” (which occupies primarily the Southeast part of the United States) into the Midwest, such that by 2050 it will occupy the entire southeastern portion of the country and all of California. The nonlinear model predicts that the zone of elevated stone risk currently located in the Southeast will expand northward to include a band of states from Kansas to Virginia and Northern California, but with the increase in prevalence primarily concentrated south of the temperature threshold. Heat exposure and dehydration constitute occupational risk factors for stone disease as well. Cooks and engineering room personnel, both of whom are exposed to high temperatures, were found to have the highest rates of stone formation among personnel of the Royal Navy (Blacklock, 1969). Likewise, Atan and colleagues (2005) found a significantly higher incidence of stones among steel workers exposed to high temperatures (8%) compared with those working in normal temperatures (0.9%). Metabolic evaluation of these two groups of workers showed a higher incidence of low urine volume and hypocitruria among the workers in the hot area. Borghi and colleagues (1993) also noted differences in the incidence of stone disease and urinary stone risk factors between workers at a glass plant who were or were not chronically exposed to high temperatures causing massive perspiration. Those exposed to high temperatures exhibited lower urine volumes and pH, higher uric acid levels, and higher urine specific gravity, leading to higher urinary saturation of uric acid. Accordingly, those workers who formed stones had a remarkably high incidence of uric acid stones (38%). Individuals with sedentary occupations such as those in managerial or professional positions have been found to carry an increased risk of stone formation for unclear reasons (Blacklock, 1969). This finding is consistent with the work of Robertson and colleagues (1980), who reported an increased risk of stone disease in affluent individuals, countries, and societies, which may be reflective of a more indulgent diet and lifestyle. The association of body size and incidence of stone disease has been investigated. In two large prospective cohort studies of men and women, the prevalence and incident risk of stone disease were directly correlated with weight and body mass index (BMI) in both sexes, although the magnitude of the association was greater in women than men (Curhan et al, 1998; Taylor et al, 2005). Although these investigators identified a reduced risk of incident stone formation with high fluid (men and women) and low protein intake (men) (Curhan et al, 1993, 1997), they found that obesity and weight gain were independent risk factors for incident stone formation and could not be accounted for by diet alone (Taylor et al, 2005). Recent evidence linking obesity and insulin resistance with low urine pH and uric acid stones (Maalouf et al, 2004a, 2004b), as well as an association between hyperinsulinemia and hypercalciuria (Kerstetter et al, 1991; Shimamoto et al, 1995; Nowicki et al, 1998), could account for an increased risk of uric acid and/or calcium stones in obese patients. A study of stone-forming and non–stone-forming participants in the Health Professionals Follow-Up Study (599 stone-forming and 404 non–stone-forming men), Nurses’ Health Study (888 stone-forming and 398 non–stone-forming older women), and Nurses’ Health Study II (689 stone-forming and 295 non–stone-forming younger women) for whom 24-hour urine studies were collected correlated urinary stone risk profiles with BMI (Taylor et al, 2006). Subjects with higher BMI excreted more urinary oxalate, uric acid, sodium, and phosphorus than those with lower BMI. Furthermore, similar to other studies, urinary supersaturation of uric acid increased with BMI. It has been suggested that the association of obesity with calcium oxalate stone formation is primarily due to increased excretion of promoters of stone formation (Siener et al, 2004; Negri et al, 2007). In contrast, the association of obesity and uric acid stone formation is primarily influenced by urinary pH. The beneficial effect of a high fluid intake on stone prevention has long been recognized. In two large observational studies, fluid intake was found to be inversely related to the risk of incident kidney stone formation (Curhan et al, 1993, 1997). Furthermore, in a prospective, randomized trial assessing the effect of fluid intake on stone recurrence among first-time idiopathic calcium stone formers, urine volume was significantly higher in the group assigned to a high fluid intake compared with the control group receiving no recommendations, and, accordingly, stone recurrence rates were significantly lower (12% vs. 27%, respectively) (Borghi et al, 1996). Geographic differences in the incidence of stone disease have been ascribed in some cases to differences in the mineral and electrolyte content of water in different areas. Although several investigators reported a lower incidence of stone disease in geographic regions with a “hard” water supply compared with a “soft” water supply, where water “hardness” is determined by content of calcium carbonate (Churchill et al, 1978; Sierakowski et al, 1979), others found no difference. Schwartz and colleagues (2002) found no association between water hardness and incidence of stone episodes, although they did observe a correlation between water hardness and urinary magnesium, calcium, and citrate levels. The solubility product and the formation product differentiate the three major states of saturation in urine: undersaturated, metastable, and unstable (Fig. 45–2). Below the solubility product, crystals will not form under any circumstances and dissolution of crystals is theoretically possible. At concentrations above the formation product, the solution is unstable and crystals will form. In the metastable range, between the solubility product and the formation product and in which the concentration products of most common stone components reside, spontaneous nucleation or precipitation does not occur despite urine that is supersaturated. It is in this area that modulation of factors controlling stone formation can take place and therapeutic intervention is directed. (From Meyer JL. Physicochemistry of stone formation. In: Resnick MI, Pak CYC, editors. Urolithiasis: a medical and surgical reference. Philadelphia: WB Saunders; 1990. p. 11–34.) To estimate the state of saturation for any given crystal system such as calcium oxalate or calcium phosphate, Pak and Chu (1973) developed a mathematic formula, the activity product ratio, that takes into account urine pH and the ionic activities of all major ion species directly involved in the stone-forming process or those that affect the overall ionic strength of the urine. Finlayson subsequently developed a computer program to measure the state of saturation, EQUIL 2, which is commonly used today (Werness et al, 1985). The relative saturation ratio or concentration product ratio is defined as the ratio of the concentration product of the urine to the solubility product of the specified stone-forming salt. A reduction in the numerator will lead to undersaturation of the urine with respect to the stone-forming salt and consequently reduce the likelihood of precipitation. This can be accomplished by reducing the urinary concentrations of the stone components (e.g., calcium or oxalate), by reducing the filtered load, or by increasing urinary reabsorption. In addition, complexation with substances such as citrate reduce available free ionic calcium and decrease the relative saturation ratio. On the other hand, manipulation of factors such as pH can significantly impact the concentration of ions such as phosphate, the generation of which is highly pH dependent. Manipulation of pH has little effect on oxalate concentration, however, because oxalic acid is a strong acid (pK = 4) and pH changes within the physiologic range will have little effect on oxalate concentration. Rodgers and colleagues (2006) recently introduced a new computer program JESS (Joint Expert Speciation System) to calculate urinary saturation of stone-forming salts as an estimate of the propensity for stone formation, thereby challenging the accuracy of the widely accepted EQUIL 2 computer program. The JESS program recognizes several soluble complexes not taken into account by EQUIL 2 including dicalcium-dihydrogen phosphate and calcium phosphocitrate, whose formation depends on pH and citrate. Consequently, the fraction of ionized calcium, phosphate, and oxalate estimated by JESS will be lower than that estimated by EQUIL 2. In order to resolve the discrepancy between the two programs, the supersaturation index (SI) according to JESS and the relative saturation ratio (RSR) according to EQUIL 2 were compared with experimentally determined urinary saturation of brushite (Pak et al, 2009b) and calcium oxalate (Pak et al, 2009a). The experimentally determined method measures the concentration product ratio (CPR) without using computer-derived ionic activities. By determining the concentration product before and after incubation with a synthetic stone-forming salt, this method directly estimates saturation by measuring the extent of stone growth (in a supersaturated solution) or dissolution (in an undersaturated solution). No significant difference was found between experimentally determined CPR and JESS-derived SI, for either brushite or calcium oxalate. However, EQUIL 2–derived RSR was consistently and significantly higher than both CPR and SI, overestimating CPR by about 80% for brushite and 50% for calcium oxalate. Because CPR is too labor intensive for routine use, saturation index according to JESS probably provides a more reliable estimation of urinary saturation than relative saturation ratio derived from EQUIL 2. Historically, urinary oxalate has been considered a more important contributor to calcium oxalate stone formation than urinary calcium (Nordin et al, 1972; Robertson and Peacock, 1980). This assumption is based on the findings of Nordin and colleagues (1972), who noted that a rise in urinary calcium concentration had less of an effect than a rise in oxalate concentration in increasing urinary saturation of calcium oxalate. They further showed that at high urinary calcium concentrations the saturation of calcium oxalate reached a plateau that did not exceed the theoretic formation product of calcium oxalate, whereas high oxalate concentrations did, thereby increasing the risk of calcium oxalate crystal formation. Pak and colleagues (2004), however, challenged the notion that urinary oxalate exerts a greater pathogenetic role than calcium in calcium oxalate stone formation. They demonstrated that the choice of stability constant used for calculating the relative saturation ratio determines the relative effects of urinary calcium and oxalate concentration. Using the commonly accepted stability constant of 2.746 × 103 (used in the EQUIL 2 program), the effect of urinary calcium and oxalate proved to be equivalent. Thus they concluded that urinary calcium and oxalate are both important and equal contributors to calcium oxalate stone formation. As such, reduction in both calcium and oxalate will be effective in reducing the relative saturation ratio, and intervention to prevent stone formation can be directed at either. When these studies were repeated using JESS, the same finding of an equivalent effect of calcium and oxalate on urinary SI of calcium oxalate was found, although the dependence of SI on calcium and oxalate was less marked than was demonstrated for RSR (Pak et al, 2009a). Homogeneous nucleation is the process by which nuclei form in pure solution. Nuclei are the earliest crystal structures that will not dissolve. Small nuclei are unstable; below a critical size threshold, dissolution of the crystal is favored over crystal growth. If the driving force (supersaturation level) and the stability of the nuclei are adequate and the lag time to nucleation is sufficiently short compared with the transit time of urine through the nephron, the nuclei will persist. Inhibitors, like citrate, destabilize nuclei, whereas promoters stabilize nuclei by providing a surface with a binding site that accommodates the crystal structure of the nucleus. In urine, crystal nuclei usually form through heterogeneous nucleation by adsorption onto existing surfaces of epithelial cells (Umekawa et al, 2001), cell debris (Fasano et al, 2001), or other crystals (Kok, 1997). Within the time frame of transit of urine through the nephron, estimated at 5 to 7 minutes, crystals cannot grow to reach a size sufficient to occlude the tubular lumen. However, if enough nuclei form and grow, aggregation of the crystals will form larger particles within minutes that can occlude the tubular lumen. Inhibitors can prevent the process of crystal growth or aggregation. Magnesium and citrate inhibit crystal aggregation. Nephrocalcin, an acidic glycoprotein made in the kidney, inhibits calcium oxalate nucleation, growth, and aggregation (Nakagawa et al, 1987; Asplin et al, 1991). Tamm-Horsfall mucoprotein, the most abundant protein in urine, inhibits aggregation (Hess et al, 1991), and uropontin inhibits crystal growth (Shiraga et al, 1992). Bikunin, the light chain of inter-α inhibitor, has been shown to be an efficient inhibitor of crystal nucleation and aggregation. Opposing views regarding the formation and growth of crystal particles have led to controversy over the concept of free crystal particle growth versus fixed particle growth. Although it was initially concluded that free particle stone formation was impossible within the normal transit time through the nephron (Finlayson et al, 1978b), later recalculation using current nephron dimensions, supersaturation, and crystal growth rates determined that crystalline particles can be formed that are large enough to be retained during normal transit time through the kidney (Kok and Khan, 1994). Fixed particle growth theory presupposes an anchoring site to which crystals bind, thereby prolonging the time the crystals are exposed to supersaturated urine and facilitating crystal growth and aggregation. A number of mechanisms have been proposed to account for crystal fixation. One favored theory proposes that oxalate-induced injury to renal tubular epithelial cells promotes adherence of calcium oxalate crystals (Miller et al, 2000). In animal models of stone formation in which administration of high oxalate loads leads to calcium oxalate crystal formation, elevated urinary levels of enzyme markers of cell injury including N-acetyl-β-glucosidase and alkaline phosphatase provide evidence of damage to renal tubular epithelial cells (Khan et al, 1992; Thamilselvan and Khan, 1998). Although the exact mechanism of oxalate-induced cell injury is not known, several studies have suggested that it is mediated by free radical formation (Thamilselvan and Khan, 1998, 1999). Not only are high concentrations of oxalate toxic to renal tubular cells, but calcium oxalate crystals themselves have also been shown to promote damage to cells (Khan et al, 1993, 1999; Thamilselvan and Khan, 1998; Thamilselvan et al, 1999). Despite these findings in animal models and in vitro systems, evidence for oxalate-induced tubular damage in humans has been lacking. Indeed, no increase in markers of oxidative stress or renal cell injury has been observed in normal individuals or stone formers after ingestion of a large oxalate load (Knight et al, 2007). How oxalate-induced renal tubular cell damage potentially promotes crystal retention is not known. Randall (1937) first observed areas of damage associated with subepithelial plaques on the renal papillae. Later, structural analyses in hyperoxaluric rats demonstrated crystals attached to the injured epithelium lining the collecting ducts (Khan, 1991). In vitro studies confirmed increased binding of calcium oxalate crystals to injured renal epithelial cells in culture (Verkoelen et al, 1998). Whether the renal tubular cells or the interstitium constitutes the primary site of stone formation is unclear. Evidence of endocytosis of calcium oxalate crystals into renal tubular cells has been demonstrated in patients with disorders of oxalate metabolism (Saxon et al, 1974; Mandell et al, 1980; Lieske et al, 1992). Intracellular incorporation of these crystals could potentially lead to cell death and deposition of crystals in the interstitium, or transport of the crystals from the luminal to the basement membrane side could promote cell damage and subsequent erosion through to the papillary surface. Knoll and colleagues (2004) demonstrated in cell culture that oxalate-induced damage was more pronounced in renal nontubular compared with tubular cell lines and, further, that renal epithelial cells were more vulnerable to the toxic effects of oxalate on their basolateral side compared with their apical (luminal) side, implicating the interstitium as a possible site of primary stone formation. In light of these recent findings, a number of investigators have revisited the role of Randall’s plaques in the pathogenesis of stone formation. Low and Stoller (1997) mapped the papillae of patients undergoing endoscopic stone removal, as well as control subjects undergoing endoscopy for unrelated reasons, and found that papillary plaques occurred in 74% of stone formers compared with only 43% of control subjects. Stoller and colleagues (2004) hypothesized that the inciting event in the pathogenesis of stones may be vascular injury to the vasa recta near the renal papilla. Repair of damaged vessel walls could involve an atherosclerotic-like reaction that results in calcification of the endothelial wall, followed by erosion into the papillary interstitium and then into the collecting ducts, where it could serve as a nidus for stone formation. Evan and colleagues (2003) presented an alternative view of the pathogenesis of stone formation on the basis of extensive analysis of papillary plaques derived from biopsies obtained during percutaneous nephrolithotomy in idiopathic calcium oxalate stone formers. They localized the origin of the plaque to the basement membrane of the thin limbs of the loops of Henle and demonstrated that the plaque subsequently extends through the medullary interstitium to a subepithelial location (Fig. 45–3). Once the plaque erodes through the urothelium, it is thought to constitute a stable, anchored surface on which calcium oxalate crystals can nucleate and grow as attached stones. Among idiopathic calcium oxalate stone formers, the volume of papillary surface covered by plaque was shown to correlate negatively with urine volume and positively with hypercalciuria (Kuo et al, 2003a, 2003b) and the number of stones formed (Kim et al, 2005a), providing further corroborating clinical evidence for this sequence of events. Furthermore, Matlaga and colleagues (2006) observed that in approximately half of a studied cohort of calcium oxalate stone formers the stones were observed to be attached to the renal papillae, suggesting that formation of attached stones is an early step in the process of stone formation. (From Evan AP, Lingeman JE, Coe FL, et al. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest 2003;111:607–16.) Using high-resolution Fourier transform infrared microspectroscopy and electron diffraction, the crystal component of plaque was determined to be calcium apatite (Evan, 2003). Further analysis revealed that the deposits consisted of individual laminated particles with mineral and organic layers. All crystals were coated with organic material, and osteopontin was indentified on the outer surface of the crystal at the junction of the overlying organic molecular layer, potentially implicating osteopontin in plaque biology (Evan et al, 2005). Daudon and colleagues analyzed more than 5000 stones associated with Randall plaques and also found that carbapatite constituted the main component of the plaque in nearly all cases (Daudon et al, 2007). One intriguing but unproven hypothesis for the origin of the calcium phosphate particles described earlier involves nanobacteria, or calcifying nanoparticles (CNPs), which are self-propogating entities that precipitate calcium apatite on their exterior membrane but for which to date no genomic material has been identified (Kajander et al, 1998). Although the existence of these particles has been questioned (Cisar et al, 2000), several lines of evidence support a role of CNPs in stone formation (Kajander et al, 2001). CNPs have been detected in blood, blood products, and kidney stones, as well as in other pathologic calcifications (Ciftçioğlu et al, 1999, 2006). They have been shown to promote rapid precipitation of calcium phosphate from blood under physiologically unfavorable conditions (Kajander et al, 1998), and in an animal model intrarenal injection of CNP was shown to induce renal calcification (García et al, 2000). In a recent study in which renal papillary and blood samples were obtained from human patients undergoing nephrectomy, immunohistochemical staining using anti-CNP antibodies was positive in 8 of 11 papillary samples in which Randall plaques were visualized and in only one of those for which they were not (Ciftcioglu et al, 2008). In addition, 12 of 14 samples positive for CNP-like spheres on scanning electron microscopy demonstrated CNP growth in culture, compared with only one of three SEM-negative samples. Although not necessarily causative, these findings loosely suggest an association between CNP and Randall plaque. The pathogenesis of stone formation in other calcium stone formers and in noncalcium stone formers may differ from that of typical idiopathic calcium oxalate stone formers. Indeed, distinct morphologic subtypes characterizing particular patient phenotypes have been found, consistent with divergent underlying pathophysiologic abnormalities. Unlike idiopathic calcium oxalate stone formers, patients with enteric hyperoxaluria due to intestinal bypass for obesity demonstrate no plaque but instead show apatite crystal deposits plugging the inner medullary collecting duct lumens, along with associated epithelial cell damage with interstitial inflammation and fibrosis (Fig. 45–4) (Evan et al, 2003). Interestingly, despite the acidic urine typically found in these patients, the crystal deposits are composed of apatite, which is generally unstable at low urine pH, suggesting discordance between local tubular pH and final urinary pH (Evan et al, 2006). (From Evan AP, Lingeman JE, Coe FL, et al. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest 2003;111:607–16.) Brushite stone formers have been found to have pathology intermediate between idiopathic calcium oxalate stone formers and intestinal bypass patients, demonstrating interstitial apatite plaque and apatite plugging of the inner medullary and terminal collecting ducts, along with associated collecting duct injury and interstitial fibrosis (Fig. 45–5) (Evan et al, 2005). The pathogenesis of brushite stones has been postulated by Evan and colleagues (2005) to occur by way of crystallization of apatite in the collecting ducts leading to collecting duct injury, cell death, and enlargement of collecting ducts. Interstitial inflammation in response to the injured cells may lead finally to progressive involvement of adjacent renal tissue. (From Evan AP, Lingeman JE, Coe FL, et al. Crystal-associated nephropathy in patients with brushite nephrolithiasis. Kidney Int 2005;67:576–91.) With distal renal tubular acidosis (RTA), patients typically exhibit extensive renal calcifications. In a subgroup of patients with distal RTA in whom most of the calcifications were surgically removable, endoscopic inspection of the renal papillae demonstrated a variety of findings (Evan et al, 2007). In some patients minimal papillary changes were observed, whereas in others the papilla were pitted and contained calcium phosphate plugs protruding from dilated collecting ducts with extensive surrounding fibrosis. Randall plaques were rarely encountered. Finally, although patients with cystinuria were found to have plugging of the terminal collecting ducts of Bellini with masses of cystine crystals, surprisingly apatite deposits were also identified in the inner medullary collecting ducts and in the thin ascending limbs of the loops of Henle. It was speculated that perhaps the alkali load associated with treatment of cystine stone formers or obstruction of the inner medullary collecting ducts by the cystine plugs resulting in an acidification defect could promote apatite crystallization (Evan et al, 2006). Whether tubular or interstitial calcifications are primarily responsible for renal stone formation is not entirely clear, and it is likely that both mechanisms play a role in particular clinical scenarios. Retention of crystals in the tubular lumen may lead to nephrocalcinosis, which may or may not be associated with renal stone formation. However, these crystals may lead to obstruction-induced tubulopathy and ultimately renal failure. Experimental evidence suggests that crystals bind preferentially to regenerating/redifferentiating renal tubular cells (Verkoelen et al, 2007). Crystal binding to the surface of these cells is thought to be mediated by a number of luminal membrane molecules including hyalurionic acid, osteopontin, annexin-II, and nucleolin-related protein. Clinical observation of nephrocalcinosis in 60% of preterm infants may be related to exposure of differentiating renal tubular epithelial cells to crystalluria caused by furosemide treatment (Ezzedeen et al, 1988; Downing et al, 1992). Indeed, the kidneys of these infants have been shown to strongly express hyaluronic acid and osteopontin at the luminal membrane (Verhulst et al, 2005). Whole urine, when added to a solution of calcium phosphate, raises the supersaturation level required to initiate calcium phosphate crystallization (formation product) (Fleisch et al, 1962). Inorganic pyrophosphate was found to be responsible for 25% to 50% of the inhibitory activity of whole urine against calcium phosphate crystallization. Using different methodology, citrate, magnesium, and pyrophosphate together were noted to account for approximately 20% of the inhibitory activity of whole urine, with citrate comprising the most important factor of the three (Bisaz et al, 1978). Citrate acts as an inhibitor of calcium oxalate and calcium phosphate stone formation by a variety of actions. First, it complexes with calcium, thereby reducing the availability of ionic calcium to interact with oxalate or phosphate (Meyer et al, 1975; Pak et al, 1982). Second, it directly inhibits the spontaneous precipitation of calcium oxalate (Nicar et al, 1987) and prevents the agglomeration of calcium oxalate crystals (Kok et al, 1986). Although it has limited inhibitory effect on calcium oxalate crystal growth, it has potent activity in reducing calcium phosphate growth (Meyer et al, 1975). Lastly, citrate prevents heterogeneous nucleation of calcium oxalate by monosodium urate (Pak and Peterson, 1986). The inhibitory activity of magnesium is derived from its complexation with oxalate, which reduces ionic oxalate concentration and calcium oxalate supersaturation (Meyer et al, 1975). In addition, magnesium reduces the rate of calcium oxalate crystal growth in vitro (Desmars et al, 1973). Polyanions including glycosaminoglycans, acid mucopolysaccharides, and RNA have been shown to inhibit crystal nucleation and growth. Among the glycosaminoglycans, heparin sulfate interacts most strongly with calcium oxalate monohydrate crystals (Yamaguchi et al, 1993). Two urinary glycoproteins, nephrocalcin and Tamm-Horsfall glycoprotein, are potent inhibitors of calcium oxalate monohydrate crystal aggregation (Nakagawa et al, 1987). Nephrocalcin is an acidic glycoprotein containing predominantly acidic amino acids that is synthesized in the proximal renal tubules and the thick ascending limb. In simple solution, nephrocalcin strongly inhibits the growth of calcium oxalate monohydrate crystals (Nakagawa et al, 1987). Nephrocalcin has been identified in four isoforms: non–stone formers excrete greater quantities of two isoforms associated with the most inhibitory activity, whereas stone formers excrete urine enriched for the two isoforms lacking inhibitory activity (Nakagawa, 1997). The isoforms with inhibitory activity were found to contain γ-carboxyglutamic acid residues that were lacking in the isoforms isolated from stone formers. Tamm-Horsfall protein is expressed by renal epithelial cells in the thick ascending limb and the distal convoluted tubule as a membrane-anchored protein that is released into the urine after cleavage of the anchoring site by phospholipases or proteases. Tamm-Horsfall is the most abundant protein found in the urine and a potent inhibitor of calcium oxalate monohydrate crystal aggregation, but not growth. The role of Tamm-Horsfall in stone formation is controversial and may depend on the state of the molecule itself, which can self-aggregate under certain conditions. A recent study using a Tamm-Horsfall (THP) knockout mouse model demonstrated spontaneous formation of calcium oxalate crystals in the kidneys of mice fed ethylene glycol and vitamin D, suggesting a protective role of Tamm-Horsfall protein against crystallization of calcium salts (Mo et al, 2004). Osteopontin, or uropontin, is an acidic phosphorylated glycoprotein expressed in bone matrix and renal epithelial cells of the ascending limb of the loop of Henle and the distal tubule. Osteopontin has been shown to inhibit nucleation, growth, and aggregation of calcium oxalate crystals, as well as to reduce binding of crystals to renal epithelial cells in vitro (Asplin et al, 1998; Wesson et al, 1998). In an osteopontin knockout mouse model, intratubular calcium oxalate crystals can be induced in mice exposed to high levels of oxalate by ethylene glycol feeding (Wesson et al, 2003). Interestingly, in a THP knockout mouse model, mice fed ethylene glycol and vitamin D exhibited a dramatic increase in osteopontin levels over baseline but still formed calcium oxalate crystals (Mo et al, 2004). The authors concluded that osteopontin may constitute an inducible inhibitor of calcium oxalate crystallization that works in conjunction with constitutively expressed Tamm-Horsfall protein to prevent crystallization. Lastly, inter-α-trypsin is a glycoprotein synthesized in the liver that is composed of three polypeptides (two heavy chains and one light chain), of which bikunin comprises the light chain. Bikunin is a strong inhibitor of calcium oxalate crystallization, aggregation, and growth in vitro (Hochstrasser et al, 1984; Atmani et al, 1999), and its expression has been shown to be upregulated in a rat model when exposed to oxalate. Renal calculi consist of both crystalline and noncrystalline components. The noncrystalline component is termed matrix, which typically accounts for about 2.5% of the weight of the stone (Boyce and Garvey, 1956). In some cases, matrix comprises the majority of the stone (up to 65%), usually in association with chronic urinary tract infection (Boyce and Garvey, 1956; Allen and Spence, 1966). The exact composition of matrix is difficult to ascertain because only 25% of it is soluble (Ryall, 1993); however, chemical analysis reveals a heterogeneous mixture consisting of 65% protein, 9% nonamino sugars, 5% glucosamine, 10% bound water, and 12% organic ash (Boyce, 1968). Among the proteins incorporated into the matrix substance are Tamm-Horsfall protein, nephrocalcin, a γ-carboxyglutamic acid–rich protein, renal lithostathine, albumin, glycosaminoglycans, free carbohydrates, and a mucoprotein called matrix substance A (Hess et al, 1996). Boyce and colleagues (1962) found that substance A is immunologically unique and present in the matrix component of all stone formers. Moore and Gowland (1975) determined that substance A is composed of three or four distinct antigens unique to stones that were detected in the urine of 85% of stone formers but in no normal individuals. A recent study using reversed-phase, high-performance liquid chromatography and tandem mass spectrometry to evaluate calcium oxalate stones identified 68 distinct proteins with 95% confidence including a significant number of inflammatory proteins (immunoglobulins, defensin-3, clusterin, complement C3a, kininogen, and fibrinogen (Canales et al, 2008). The exact role of matrix in stone formation, whether as a promoter, an inhibitor, or a passive bystander, has yet to be elucidated. Thirty to 40 percent of dietary calcium is absorbed from the intestine, with most being absorbed in the small intestine and only approximately 10% absorbed in the colon (Bronner et al, 1999). By a process of intestinal adaptation, absorption of calcium varies with calcium intake. At times of low calcium intake, fractional calcium absorption is enhanced; during high calcium intake, fractional calcium absorption is reduced. With a calcium-rich diet, a nonsaturable, paracellular pathway for calcium absorption predominates. A saturable, vitamin D–dependent transcellular pathway constitutes the major pathway for intestinal calcium absorption when calcium intake is limited; this pathway is downregulated by a diet replete in calcium (Buckley et al, 1980; Bronner et al, 1986). Because of the saturable component of calcium transport, a larger portion of calcium is absorbed when it is divided into several doses taken hours apart compared with a large single dose (Phang et al, 1968). A small amount of calcium is secreted into the lumen of the intestine, thereby reducing net calcium absorption, such that, overall, 100 to 300 mg of a total average calcium intake of 600 to 1200 mg daily will be absorbed. Key Points: Physicochemistry Calcium is absorbed in the ionic state, and incomplete calcium absorption is due in part to formation of soluble calcium complexes in the intestinal lumen. As such, substances that complex calcium such as phosphate, citrate, oxalate, sulfate, and fatty acids reduce the availability of ionic calcium for absorption (Allen, 1982). Calcium readily complexes with phosphate in the intestinal lumen, but because calcium phosphate formation is dependent on pH (pK = 6.1), high luminal pH favors calcium phosphate complexation, thereby reducing calcium availability. On the other hand, calcium oxalate complex formation displays less pH dependence and complex formation is less reversible. Consequently, an oxalate-rich diet reduces calcium absorption. Transcellular calcium absorption is mediated by 1,25(OH)2D3 (calcitriol), which is reported to enhance calcium permeability at the brush border of the intestinal epithelial cells (Fontaine et al, 1981). Calcitriol also acts on the bone and kidney in addition to its action in increasing calcium absorption from the intestine. In the bone, 1,25(OH)2D3, along with PTH, promotes the recruitment and differentiation of osteoclasts that subsequently mobilize calcium from the bone. Consequently, the filtered load of calcium and phosphate increases. However, PTH increases renal calcium reabsorption and enhances phosphate excretion, leading to a net increase in serum calcium, which ultimately suppresses further PTH secretion and synthesis of 1,25(OH)2D3. Calcitriol modulates parathyroid function as well by inhibiting synthesis of PTH through enhanced vitamin D receptor and calcium-sensing receptor expression in the parathyroid glands (Dusso et al, 2005). PTH is critical in maintaining normal calcium concentration in the extracellular fluid. PTH is an 84 amino acid protein that is the cleavage product of the precursor protein preproPTH. Only mature PTH is secreted from the parathyroid gland, and the most potent stimulus for its secretion is a decrease in serum calcium (Sherwood et al, 1968). In response to serum calcium levels, the G-protein-coupled extracellular calcium-sensing receptor (CaSR) regulates PTH secretion and renal tubular calcium reabsorption (Devuyst and Pirson, 2007). PTH stimulates mobilization of calcium from bone through the action of osteoclasts, further raising serum calcium and phosphorus. The action of PTH is mediated through changes in cyclic adenosine monophosphate (AMP) and phospholipase C (Dunlay et al, 1990; Muff et al, 1992). At the kidney, PTH enhances renal calcium reabsorption and reduces renal tubular reabsorption of phosphate.
Epidemiology of Renal Calculi
Gender
Race/Ethnicity
Age
Geography
Climate
Occupation
Body Mass Index and Weight
Water
Physicochemistry
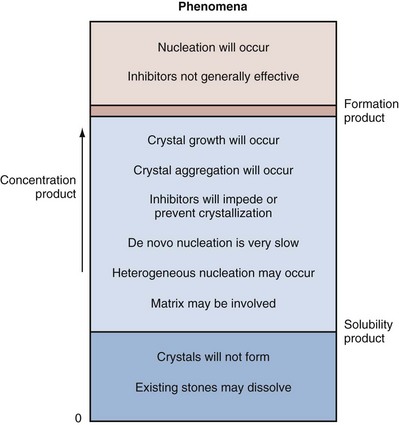
State of Saturation
Nucleation and Crystal Growth, Aggregation, and Retention
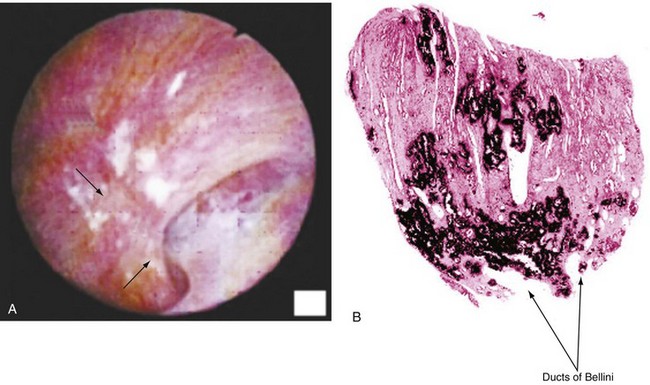
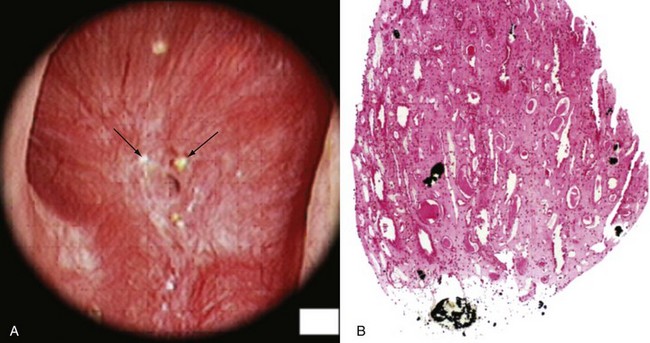
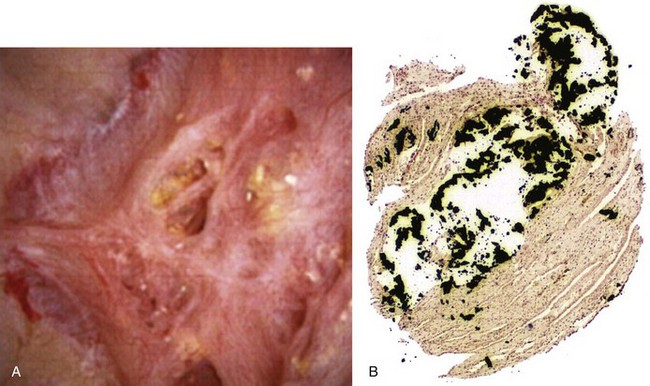
Inhibitors and Promoters of Crystal Formation
Matrix
Mineral Metabolism
Calcium
Related posts:
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Surgery for Bladder Cancer
Surgery for Bladder Cancer
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Urinary Lithiasis: Etiology, Epidemiology, and Pathogenesis
