1
Antiphospholipid syndrome
2
Systemic lupus erythematosus
3
Systemic scleroderma
4
Malignant hypertension
5
Pregnancy-HELLP syndrome
6
Drugs
7
Malignancy
8
Solid organ transplant
9
Allogeneic stem cell transplant—ionizing radiation
10
Infections: Sepsis, HIV
11
Pancreatitis
12
Surgeries
Microangiopathic Antiphospholipid-Associated Syndrome-Associated TMA
Antiphospholipid syndrome (APS) is an autoimmune disorder characterized by the persistent presence of circulating antiphospholipid antibodies (aPL) and vascular (arterial and/or venous) thrombosis and/or pregnancy-related complications. Antiphospholipid antibodies are a group of antibodies directed against plasma proteins that possess ionic phospholipids, primarily β2-glycoprotein I (β2GPI), prothrombin, and phosphatidylserine–prothrombin complex (PS/PT). In 2006, the definition of APS was updated, and thrombotic conditions affecting mainly microvasculatures (without large-vessel thrombosis or occlusions), i.e., TMA-associated conditions in patients with aPL positivity, were included in the disease spectrum and, hence, microangiopathic APS (MAPS) [1, 2].
The precise pathogenesis of TMA in MAPS has not been fully elucidated. Clinically, despite the persistent presence of circulating antiphospholipid antibodies, thrombotic events occur only occasionally, suggesting other factors are needed to trigger thrombotic events. There has been a proposal of a “two-hit hypothesis” in which aPL, representing the first hit, causes thrombotic events in the presence of another thrombophilic stimulus, the second hit. The two-hit hypothesis is supported by an animal model of APS in which aPL injection to rats resulted in thrombus formation only when the animals were also pretreated with lipopolysaccharide, not with normal saline [3].
Although not fully elucidated, aPL are thrombogenic via three potential mechanisms: (1) diminished effects of nitric oxide (NO); (2) abnormal interactions of aPL with coagulation factors and cells associated with the regulation of thrombosis, mainly monocytes, endothelial cells, and platelets; and (3) aberrant complement activation.
Nitric oxide, generated by the enzymatic action of endothelial nitric oxide synthase (eNOS), is a key regulatory molecule in maintaining vascular health. Nitric oxide regulates the expression of adhesion molecules and inhibits platelet aggregation by augmenting cGMP production in platelets. There are a number of studies in both mouse models and humans suggesting a role for impaired NO production and activity in the genesis of TMA. aPL binding to β2GPI is shown to inhibit eNOS activation with a resultant deficiency of NO production which in turn predisposes to a full-scale leukocyte (monocyte)-endothelium adhesion and thrombus formation [4], especially when there is a second hit.
aPL is directed against plasma protein β2GPI; binding of aPL and β2GPI increases the affinity of aPL to membrane phospholipids. The interaction of aPL and membrane of platelets, monocytes, and endothelial cells can activate prothrombogenic cellular elements, promoting thrombogenesis. Studies have shown that the interaction of aPL and cell membrane requires co-activation of specific receptors. A number of candidate receptors have been implicated, including annexin A2, glycoprotein Ibα and apolipoprotein E receptor 2, toll-like receptors (TLR-2 and TLR-4), VLDL receptor, and possibly other receptor proteins [5–7].
Under physiological condition, β2GPI binds to the membrane of activated platelets and inhibits the generation of activated factor X. The binding of aPL and β2GPI interferes with such inhibition and promotes platelet aggregation and thrombus formation [8]. The binding of aPL to the membrane of monocytes induces upregulation of tissue factors, a hallmark of monocyte activation, also seen in patients with APS [9]. The binding also activates signaling cascades including, but not limited to, P38 MAPK-mediated activation of tumor necrosis factor (TNF) alpha, IL-1b and macrophage inflammatory cytokine 3b, NFκB, and upregulation of procoagulant substances and adhesion molecules [10, 11]. In the endothelium, pathogenic binding of aPL causes an upregulation of myeloid differentiation protein (MyD)88-dependent signaling, adhesion molecules, tissue factors, and endothelin-1, resulting in a pro-inflammatory and prothrombogenic endothelial cell phenotype [12]. The binding also stimulates endothelial cells to release microparticles that likely promote a thrombogenic endothelial cell phenotype [10, 11, 13].
Recently, complement activation has emerged as one of the important mechanisms related to the development of TMA in patients with MAPS [14–16]. Hypocomplementemia, caused by complement activation and consumption, is a commonly observed feature of MAPS. Activation of complement pathway leads to C3 activation, which induces the production of pro-inflammatory fragment of C3a (also called anaphylatoxin) and subsequent C5 activation and production of anaphylatoxin C5a, another strong inflammatory toxin. C5b, another fragment of activated C5, forms complex with C6, C7, C8, and C9 leading to the formation of a multi-complex membrane attack protein (MAC) and ultimately causing disruption of cell membrane and cell death. Thus, activation of complements produces anaphylatoxins and fragments that amplify the activation of monocytes, endothelial cells, and platelets. The complement activation also induces expression of tissue factor as well as adhesion molecules, further promoting platelet aggregation [17, 18]. The anaphylatoxin 5a is especially important in the genesis of placental damage in MAPS [19]. Obstetrical complications associated with MAPS can be markedly suppressed in animal models when complement is made deficient or anticomplement antibody is administered [20].
Histopathologically, TMA-associated MAPS shows thrombi in the capillary lumina in the kidney biopsy. It can be indistinguishable from TMA in the classic form of TTP-HUS. There could also be MAPS-related changes including fibrous intimal fibroplasia, focal cortical atrophy, and artery and/or arterial occlusions. Figure 16.1 shows biopsy images from a 42-year-old woman with MAPS.
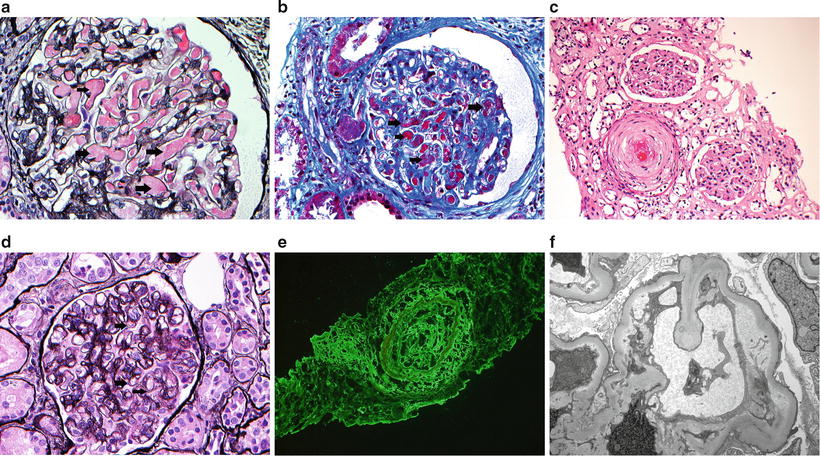
Fig. 16.1
Thrombotic microangiopathy. (a–c) Light microscopy showing thrombus within small arteries (a: silver methenamine; b: trichrome; c: hematoxylin and eosin). In c, note ischemic glomeruli around artery with thrombosis. (d) Light microscopy showing features of chronic thrombotic microangiopathy with thickened peripheral capillary walls and double contour formation (arrows) (silver methenamine). (e) Immunofluorescence microscopy showing fibrinogen within an artery. (f) Electron microscopy showing chronic thrombotic microangiopathy with subendothelial expansion with fluffy granular material and cellular elements. Note endothelial injury as manifested by loss of fenestrations
Treatment modalities for the majority of patients with MAPS include chronic anticoagulation. In cases with TMA, depending on the severity and the degrees of organ involvement, high-dose steroids, IVIG, and/or plasmapheresis may be tried [21]. Case reports also suggest a beneficial effect of rituximab, a B-cell depletion antibody, for patients with severe MAPS [22].
In patients who develop end-stage renal failure, a positive titer of circulating aPL is not a contraindication for kidney transplant. However, posttransplant patients should be on chronic anticoagulation, and calcineurin inhibitors should be avoided. When necessary, rituximab can be tried. Recently, eculizumab has been used successfully for several kidney transplant recipients with positive aPL to prevent and to treat TMA [23, 24]. Eculizumab is a humanized monoclonal antibody that inhibits C5 activation. It has shown efficacy in treating patients with paroxysmal nocturnal hemoglobinuria (PNH). One limitation with this drug is its cost, running approximately $15,000 (September 2012 USD) for a maintenance weekly dose (900 mg) and $400,000 for one year’s maintenance supply. Eculizumab is also known to increase the risk of meningococcal infections. Meningococcal vaccine is therefore required for all patients at least 2 weeks prior to beginning eculizumab therapy. Moreover, patients on eculizumab should have ongoing monitoring for early symptoms and signs of meningococcal infections to allow the start of treatment for the infections in a timely fashion. Eculizumab was recently approved in the USA as a first-line drug for atypical HUS (aHUS). However, its long-term use requires further evaluation. In patients with refractory MAPS-associated TMA, eculizumab may be tried.
Systemic Lupus Erythematosus-Associated TMA
TMA occurs in patients with a variety of connective tissue diseases. Matsuyama et al. [25] examined 127 patients with connective tissue disease-associated TMA. Among the subjects, 64 were patients with SLE, 42 with systemic sclerosis [excluded patients in scleroderma renal crisis (SRC)], 11 with polymyositis/dermatomyositis, and ten with rheumatoid arthritis. Most of the 127 patients demonstrated a normal or mild degree of deficiency in ADAMTS13 activity, which was consistent with the existing literature and would not explain the development of TMA. Only a small fraction of this group of TMA patients had severe deficiency of ADAMTS13 activity which was mostly associated with circulating neutralizing antibodies. Here, we will focus on TMA in patients with SLE.
SLE, when active, is often associated with a mild degree of anemia and thrombocytopenia. Thrombocytopenia in SLE can occur through a variety of mechanisms including antiplatelet antibody, ITP- or TTP-like syndrome, and TMA with or without aPL. Although clinical presentation of SLE-associated TMA can be indistinguishable from TMA in idiopathic/classic TTP-HUS, there are differences in its incidence, its relation to ADAMTS13 activity, and its response to plasma exchange. The incidence of SLE-associated TMA is dramatically higher than idiopathic TTP-HUS, approximately 1–4 % of SLE patients and 3.7 per million residents, respectively [26]. In 2009, a single-center study showed an incidence rate of TMA in SLE of approximately 2.2 % [26]. Moreover, studies have demonstrated that TMA in SLE is not associated with an overall trend of reduction in ADAMTS13 activity, nor with dominant neutralizing antibodies against ADAMTS13, as seen in idiopathic TTP-HUS [26]. Consequently, TMA in SLE is quite resistant to plasmapheresis, unlike the majority of idiopathic TTP that are highly responsive to such treatment. Thus, TMA in SLE likely differs etiologically, at least in part, from TMA in idiopathic TTP-HUS.
Multiple etiological factors are speculated to be in play in SLE-associated TMA, including the involvement of autoantibodies or inhibitory antibodies targeting ADAMTS13 activity (in minority of the cases), D-dimer elevation which can promote microvascular thrombus formation, reduction in NO-mediated vasodilatory effects through the binding of hemoglobin to NO and potentially ADAMTS13 inhibitory effects exerted by hemoglobin, general procoagulant state in SLE patients with and without the presence of aPL, increased activity of plasminogen activator inhibitor (PAI) leading to insufficient antithrombin III activity, underlying inflammatory state in SLE-causing resistance of vWF multimer to ADAMTS13-mediated cleavage, and direct endothelial cell activation due to multiple antibodies produced in the setting of SLE. A Scottish epidemiologic study showed that severe sepsis/infections can also be associated with the onset of TMA in patients with SLE, suggesting infection and sepsis could be a triggering event for TMA in lupus. Another study by Kwok et al. [27] showed that SLE disease activity can sometimes be a trigger for the onset of TMA in lupus. Similar results were reported by Chen et al. in 2011 [28]. Although all of the above have been proposed as contributing elements in the development of TMA in patients with SLE, the precise mechanisms are far from certain.
SLE affects multiple organ systems including the central nervous system, kidneys, bone marrow, blood elements, and aggregate cytokine release which can all mimic the presentation of idiopathic TTP, making the diagnosis of SLE-associated TMA a challenge. Providers should be on high alert for this condition, especially in patients with new onset thrombocytopenia and microscopic hemolytic anemia with a background SLE. Future standardization of tests for ADAMTS13 antigen, anti-ADAMTS13 antibody, and inhibitors of ADAMTS13 could potentially help with clarifying some of the differential diagnosis.
SLE-associated TMA carries a grim prognosis. Mortality rate in SLE patients with TMA is 34–62 % [29, 30], much higher than TMA patients without SLE, <10 % [31]. Older age, renal failure, and infections can all contribute to the higher mortality rate.
Treatment for SLE patients with TMA remains a real challenge. Strategies employed include steroids, cyclophosphamide, azathioprine, IVIG, and recently rituximab. An initial trial of plasmapheresis may be considered in cases when there is a suspicion for idiopathic TTP-HUS. In the setting of infection, the dosage of immunosuppressants should be reduced. Although neutralizing antibody does not seem to be important in SLE-associated TMA, there have been case reports of using rituximab, an inhibitor of B-cell-mediated antibody production, for treating patients with SLE-associated TMA and showing beneficial effects [32, 33]. These strategies have not been universally accepted. Better understanding the underlying mechanisms could help with development of novel approaches to its treatment.
In summary, SLE patients have a relatively high incidence of TMA compared to idiopathic TTP-HUS. Although the precise pathogenesis is not well understood, TMA in SLE does not appear to be associated with reduction in ADAMTS13 activity. TMA in SLE patients signifies poor prognosis and high mortality rate. There is no specific protocol for the treatment of TMA in patients with SLE. Future studies should be focused on clarifying the underlying pathogenesis which could potentially lead to improvement in treatment strategies and reduced mortality.
Systemic Scleroderma Renal Crisis-Associated TMA
TMA associated with systemic scleroderma (SSc) occurs typically in patients with scleroderma renal crisis (SRC) which is defined as a rapid decline in kidney function associated with hypertension without an alternative explanation. SRC develops in ~5 % of patients with systemic scleroderma, 12–19 % in diffuse systemic sclerosis and 2 % in limited systemic sclerosis [34–36]. Approximately half of the patients with SRC develop TMA.
Clinical features of SRC include (1) progressive renal impairment and often accelerated hypertension, (2) microangiopathic hemolytic anemia occurring in ~50 % of SRC patients, and (3) urinalysis showing non-nephrotic range proteinuria, hematuria, and often granular casts. Renal failure in SRC often develops over weeks rather than days. Accelerated hypertension is present in most cases but is not universal. Several reports have noted ~10–20 % normotensive SRC which usually portends a poor prognosis and high rate of irreversible renal failure and mortality. Rarely, SRC can present in association with diffuse alveolar hemorrhage along with TMA [37].
Several risk factors for SRC have been identified. Early phases of diffuse sclerosis (within months of a diagnosis of SSc) are at the greatest risk for SRC. An estimated 66 % of patients with SSc develop SRC within 1 year of the diagnosis [36, 38]. Other risk factors include rapid progressive skin lesions, recent exposure to glucocorticoids, hormone replacement therapy, and presence of SSc-specific anti-RNA polymerase antibodies. The presence of anticentromere antibody reduces the SRC risk.
In a recent multicenter retrospective study by Guillevin et al. [39] of patients with SRC, hypertension occurred in 85.7 % and hypertensive encephalopathy was found in about 60 %. More than half (56 %) of the SRC patients showed evidence of TMA. Overall, the outcome of SRC was poor. 53.8 % of patients required dialysis and 40.7 % of the patients died compared to 10.8 % in control (SSc patients without SRC). Normotensive SRC patients had worse outcomes; none of them recovered kidney function and all remained dialysis dependent. Dialysis patients showed a higher mortality rate. There was no mention of the impact of TMA on the disease presentation and outcomes in this specific study, although in the literature SRC patients with TMA typically fare worse with higher mortality rate than SRC patients without TMA.
The routine use of angiotensin-converting enzyme inhibitor (ACEi) has improved the outcomes of SRC with a reduction in 12-month mortality rate from 76 % to less than 15 % in the USA [40]. Treatment for SRC patients with or without TMA is to immediately institute ACEi and/or ARB as tolerated. Immunosuppressive regimen and/or biotherapy have not been shown to improve clinical outcomes. Further studies are needed to clarify the usefulness of these treatments.
Malignant Hypertension-Associated TMA
TMA can develop in patients with malignant hypertension which occurs in ~1 % of hypertensive patients [41]. Malignant hypertension is a clinical syndrome featuring a severely elevated blood pressure (>180/120 mmHg), encephalopathy, congestive heart failure, acute papilledema, and funduscopic hemorrhage with exudate, change of mental status, acute kidney failure with microscopic hematuria and proteinuria. TMA associated with thrombocytopenia and microscopic hemolytic anemia can be found in about a third of patients with malignant hypertension [42].
TMA in malignant hypertension is caused by fibrinoid necrosis of small arteries and arterioles. The fibrinoid necrosis leads to a narrowing of the vessel lumen resulting in red blood cell fragmentation as the cells pass through the vessels. A study by van de Born et al. shows a mild reduction of ADAMTS13 in patients with TMA associated with malignant hypertension [43]. It is suggested that reduced ADAMTS13, endothelial activation and release of endothelial VWF can work in concert in the TMA genesis. Pathologically, intrarenal TMA in patients with malignant hypertension is similar to TMA resulting from other causes. Specific features of fibrinoid necrosis in the arterioles with fine subendothelial lipid droplets and hyalin thrombi formation are prominent.
Immediate control of blood pressure is the only effective method for reversing the process of hypertensive vascular injury and rescuing kidney function. With effective blood pressure control, platelet count typically normalizes within a week [44]. Parenteral therapy using antihypertensive agents with a rapid onset of action and short drug half-life in a monitored setting is necessary to ensure a precise and optimal blood pressure control. Agents that are being used for hypertensive emergency include nitroprusside, beta-blockers, labetalol, calcium-channel antagonists (including clevidipine, a recently introduced third-generation dihydropyridine calcium-channel antagonist) [45], hydralazine, ACEi, and fenoldopam. Depending on the nature and severity of target-organ injury, one drug could be preferred over another. For instance, clevidipine is an ultra short-acting calcium-channel antagonist with a rapid onset and half-life of <1 min. It is metabolized by red blood cell esterases, independent of renal and hepatic function. Clevidipine reduces blood pressure by a direct effect on arterioles, reducing afterload without affecting cardiac filling pressure or causing reflex tachycardia. Fenoldopam is another short-acting vasodilator. It is a dopamine 1-receptor agonist that increases renal blood flow and natriuresis. Labetalol is also a rapid onset and short-acting antihypertensive that blocks alpha-1 and (nonselective) beta-adrenergic receptors. It reduces afterload without reducing cerebral, coronary, and renal blood flow. Labetalol is safe in patients with acute coronary syndrome and is one of the most commonly used intravenous antihypertensive for patients with hypertensive emergency. However, there is no standard protocol and no evidence from large randomized controlled trials to support the use of one therapeutic agent over another with regard to its efficacy [46]. Consequently, practices vary considerably.
Overall outcomes improve with control of blood pressure [41, 47]. Without effective blood pressure control, the mortality rate can be as high as 90 %. With effective blood pressure control, the kidney function can be stabilized and improved in patients with a mild degree of kidney dysfunction (baseline serum creatinine <2.0 mg/dL). In patients with advanced baseline chronic kidney dysfunction, serum creatinine level >4 mg/dL, end-stage renal failure typically ensues and permanent renal replacement therapy is necessary [48].
Pregnancy-Associated TMA
Since the first report of pregnancy-associated TMA in 1955 by Miner et al. [49], there have been a number of case reports and case series showing that pregnancy is rarely associated with the occurrence of TMA with an estimated incidence of 1:25,000 birth [50, 51]. It is important to recognize the studies predate the era of testing for ADAMTS13 activity; thus, pregnancy-associated TMA was not distinguished from the classic TTP-HUS.
Pregnancy is a risk factor for the development of TMA. This is because even under physiological condition, pregnancy is associated with increasing concentrations of coagulating factors, decreased fibrinolytic activity, and decreased activity of ADAMTS13 [52–55]. Mannucci et al. [55] showed the mean value of ADAMTS13 activity in women during the second and third trimesters was significantly lower than during the first trimester, 64 % versus 94 %. Patients heterozygous for ADAMTS13 deficiency can potentially become more severely deficient of this enzyme during pregnancy, triggering the development of TMA. In patients with underlying genetic and procoagulant defects, such as the presence of factor V Leiden, methylenetetrahydrofolate reductase (MTHFR) 667T, factor II 20 210A, and plasminogen activator inhibitor (PAI-1) 4G, and deficiencies in protein S, protein C, and antithrombin III, pregnancy can serve as a precipitating factor to trigger acute thrombotic events [56]. These underlying genetic or acquired abnormalities become progressively more severe in the course of pregnancy with the maximum abnormalities occurring at near term and immediately postpartum.
It is not surprising that pregnancy-associated TMA tends to occur at term or shortly following delivery. The onset of the TMA tends to accompany symptoms of preeclampsia, hypertension, and often prominent gastrointestinal symptoms. Severe neurological impairment and renal failure typically ensue, and death can be imminent without a timely diagnosis and treatment [50].
No firm treatment guidelines have been devised for pregnancy-associated TMA. Hayward et al. reviewed 67 episodes of TMA in 52 consecutive pregnant patients over a 12-year-period and showed that a plasma exchange-based therapy was associated with >95 % (65 of 67 cases) hematologic response. Despite the high response rate, two patients in remission were brain-dead and 8 % of the patients were dead in a median follow-up duration of 1.1 years. Persisting renal impairment, hepatitis, and transfusion-associated acquired immunodeficiency syndrome were seen in association with the disease and the treatment [57]. Other studies have also reported a substantial morbidity and maternal and fetal mortality despite treatment [58, 59].
Due to the poor prognosis of TMA during pregnancy and the potential confusion in the diagnosis of severe preeclampsia versus idiopathic TTP-HUS versus pregnancy-associated TMA, plasma infusion or plasma exchange and corticosteroid therapy are usually tried [57–59]. Although no standard treatment guidelines have been devised for pregnancy-associated TMA, plasma exchange or infusion is considered beneficial. The role of steroids for this condition is, however, unclear. Supportive measures, including blood pressure control and symptomatic management, should be optimized.
Although pregnancy likely acts as a precipitating event for acute TMA episodes, the risk for TMA in the future pregnancies in women who have experienced an episode of pregnancy-associated TMA is unclear. Clinical research is needed to evaluate the risk of subsequent pregnancies for women who have recovered from pregnancy-associated TMA, especially for those without genetic risk factors.
Drug-Associated TMA
A number of United States Food and Drug Administration (FDA)-approved drugs are known to be associated with TMA. Several herbal remedies including morning cupressus funebris and echinacea pallida and a diet remedy called chromium picolinate have also been reported to induce TMA-like symptoms including hemolytic anemia, thrombocytopenia, and acute kidney failure [60].
The pathogeneses of drug-associated TMA are multiple. Drugs can cause TMA via immune-mediated mechanism, via drug-dosage-dependent toxicity, or via unknown (yet to be elucidated) mechanisms. The only drug that has been proven to cause immune-mediated TMA is quinine. Quinine is a commonly used medication for leg cramps. Common beverages such as tonic water, Dubonnet Aperitif, and Schweppes bitter lemon also contain quinine. Quinine has been shown to induce production of specific antibodies against platelets, neutrophils, lymphocytes, and endothelial cells [61]. These antibodies are capable of inciting immune-mediated reaction, causing TMA and acute kidney failure. Additionally, they can cause neutropenia, disseminated intravascular coagulation, and liver dysfunction [62, 63].
Another group of drug-associated TMA is related to drug dosage. Dosage-dependent development of TMA has been observed mostly in chemotherapeutic agents including (in descending order) mitomycin C, gemcitabine, calcineurin inhibitors (cyclosporine and tacrolimus), carmustine, and pentostatin [64–68]. Dose reduction has been shown to ameliorate or eliminate the TMA.
There are a number of medications that have been reported to induce TMA due to unclear mechanism(s). For instance, ticlopidine and clopidogrel have been shown to occasionally induce TMA, although the underlying pathogenesis is unclear [69, 70]. Other medications in this category include deoxycoformycin, oxaliplatin, trimethoprim-sulfamethoxazole, and recently VEGF inhibitors [71–75]. Estrogen is another group of drugs that are known to trigger TMA. Oral contraceptives, hormonal replacement regimens, and tamoxifen have all been reported to induce TMA [76–78]. The underlying mechanisms might be related to a reduced production of prostacyclin [79].
Diagnosis of drug-associated TMA relies on a detailed drug-intake history and chronological association and the absence of other potential causes of TMA. Immune-mediated reaction associated with quinine tends to have acute onset and be severe, while nonimmune-mediated and drug-dosage-associated TMA tends to show a more gradual onset and presents with increasing TMA severity with cumulating drug exposure.
Treatment should focus on correct diagnosis and discontinuation of offending agents. A trial of plasma exchange may be considered in view of the potentially life-threatening nature of TMA, diagnostic confusion in differentiating drug-induced TMA from idiopathic TTP-HUS, and lack of other treatment options. Glucocorticoids are in general not used as their efficacy in drug-associated TMA has not been tested. Several strategies have been instituted for treating calcineurin-inhibitor-associated TMA with some success. These include cessation of calcineurin inhibitor, dose reduction, and switching one calcineurin inhibitor for another or replacing calcineurin inhibitor with sirolimus or belatacept. Unfortunately, sirolimus has been reported to induce posttransplant TMA itself. Some patients with cyclosporine-associated TMA can be rechallenged with tacrolimus without developing recurrent TMA. The combination of sirolimus with either cyclosporine or tacrolimus is associated with a higher incidence of TMA [80] and should be avoided. Plasma exchange has frequently been used as a part of the management for calcineurin-inhibitor-associated TMA especially for patients with multisystemic involvement of TMA. In one report, the recovery of the kidney graft function reached 80 % (23 of 29 patients) by a combination treatment of plasma exchange and discontinuation of calcineurin inhibitors [81]. Mean duration of plasma exchange therapy was 8.5 (range 5–23) days in that study. IVIG has also been used either alone or with plasma exchange or as rescue therapy and the success rate has been variable. An optimal therapy for calcineurin-associated posttransplant TMA requires randomized trials which are not currently available. For VEGF-inhibitor TMA, a recent nonrandomized study showed that plasmapheresis demonstrated no outcome advantage over conservative measures and is associated with more adverse effects [82]. Recognition of potential drug etiology as a cause of TMA is critical and foremost to avoiding future exposure to the offending drug.
Cancer-Associated TMA
TMA can be associated with virtually any type of malignancy especially disseminated cancers [64]. Most patients with cancer-associated TMA exhibit overt clinical evidence of cancer. However, in a small fraction of the patients, TMA can be the presenting manifestation of cancer or cancer recurrence [83] and, consequently, be misdiagnosed as having the classic TTP-HUS and be treated inappropriately with plasma exchange.
Postulated pathogeneses for cancer-associated TMA include microvascular obstruction due to tumor emboli, activation of coagulation, and tumor-stimulated intimal proliferation. Cancer-associated TMA does not seem to be mediated by a deficiency of ADAMTS13 activity [84, 85].
Cancer-associated TMA is often accompanied by clinical features atypical for the classic TTP-HUS. Patients with cancer-associated TMA tend to be older, have previously been diagnosed with cancer, and present with a smoldering progression of symptoms over several months rather than several weeks as in the classic TTP-HUS. These patients also experience gradual onset of weight loss, generalized weakness, pain, and respiratory symptoms such as cough and dyspnea which are rare in patients with the classic TTP-HUS. Besides the characteristic biochemical changes associated with TMA, patients with cancer tend to show a much higher level of circulating LDH compared to that in the classic TTP-HUS [84, 86]. Severe ADAMTS13 deficiency (<5 %) and/or inhibitory ADAMTS13 antibodies are typically absent [85]. Identifying these features in TMA patients should raise suspicion for potential cancer-associated TMA.
The treatment should focus on the underlying malignancy. Plasma exchange has not been beneficial in patients with cancer-mediated TMA and is not indicated. In general, prognosis for cancer-associated TMA is poor as most of the patients have late-stage malignancy. Median survival after the diagnosis of tumor-associated TMA is typically in days or weeks [84, 86].
Solid Organ Transplant-Associated TMA
TMA is a recognized complication following solid organ transplantation (SOT), with an occurrence rate of roughly 0.5–15 %. Proposed risk factors for the development of TMA include HLA incompatibility, longer duration of the transplant surgery, lack of perioperative use of fresh frozen plasma, the use of calcineurin inhibitors, and underlying preexisting hypercoagulable abnormalities. The onset of post SOT-associated TMA is usually within the first few weeks after transplantation. Clinical features include anemia, reticulocytosis, LDH elevation, positive schistocytes on peripheral blood smear, and varying degrees of kidney dysfunction. The severity of this entity varies and may range from minimally symptomatic to fulminant and widespread multiorgan dysfunction with little chance of survival.
The pathogenesis of post SOT-associated TMA is incompletely understood, although direct endothelial injury related to immunosuppressive drugs, specifically calcineurin inhibitors and sirolimus; vascular rejection of the allograft; infections specifically with CMV reactivation; and other insults occurring in the course of posttransplant surgery have all been implicated in its pathogenesis (reviewed in [87]).
TMA may complicate ~1–15 % of kidney transplants among recipients on calcineurin-inhibitor-based immunosuppressive regimen [88, 89]. Most of the posttransplant TMA cases occur during the initial period of transplantation, although occasional cases have been reported to occur for up to 6 years. About a third of the posttransplant TMA cases is limited to the kidney graft and detected by allograft biopsy [89]. The remaining cases of TMA are associated with a varying degree of systemic manifestations including thrombocytopenia, microangiopathic hemolytic anemia, and organ dysfunctions. The use of calcineurin inhibitors is thought to induce vasoconstriction, endothelial injury, and hypercoagulability, potentially predisposing to TMA. Coexisting allograft ischemia-reperfusion injury presents another risk for the potential development of TMA [90]. Although long-term pathologic evolution of acute renal TMA has not been well described, the overall allograft survival is reduced with kidney transplant-associated TMA.
Studies have shown that TMA can occur in ~8 % adult recipients of living-donor liver transplant. The underlying mechanisms are also thought to be multifactorial causing direct vascular injury. In a study of 393 liver transplant recipients, 30 patients (7.6 %) developed TMA. TMA in this group of patients was associated with a reduced survival with 1-year survival rates of 60.6 % [91].
The optimal treatment strategy for post SOT-associated TMA has not been developed. The general approach has been to eliminate all possible causative conditions and potential offending medications such as cyclosporine, tacrolimus, and sirolimus. Aggressive treatment of potential opportunistic infections is critical as death in this condition is often associated with infections and other transplantation-related complications.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


