Fig. 10.1
On ethanol-fixed neutrophils, proteinase-3 (PR3) ANCA cause a characteristic cytoplasmic granular centrally accentuated immunofluorescence pattern, referred as c-ANCA (top), while myeloperoxidase (MPO)-ANCA causes a perinuclear immunofluorescence pattern, referred to as p-ANCA (bottom)
Despite the many clinical similarities, there are substantial clinical differences between these three syndromes, and the different ANCA types portend different prognostic implications. Consequently, a nuanced clinical management approach to these patients is warranted. With emphasis on renal manifestations, this chapter aims to highlight the current understanding of the pathogenesis of AAV as well as clinical differences between the syndromes and different ANCA types as they impact treatment decisions and prognosis.
Epidemiology
Together, the AAV have a reported incidence of 10–20 cases per million per year [9]. The distribution of the specific syndromes varies geographically and with ethnicity [10]. GPA is more common in Northern than in Mediterranean Europe, where MPA is more common [10]. GPA is also exceedingly rare in African-Americans and in Japan, where it is almost exclusively encountered on the northern island of Hokkaido [11, 12]. A similar latitude-dependent incidence gradient has also been documented in New Zealand [13]. Furthermore, in New Zealand as well as in France, the incidence of AAV is higher in the populations of European ethnicity than among non-Europeans, Asians, or Pacific Islanders [13, 14].
Etiology and Pathogenesis
The etiology of the AAV syndromes remains unclear. Like for many other polygenic systemic autoimmune diseases, GPA, MPA, and EGPA are in all likelihood the result of complex interactions between genetic factors predisposing for the loss of self-tolerance and autoimmunity and triggering environmental exposures. These factors induce and maintain inappropriate lymphocyte activation and autoantibody production which lead to tissue injury and predispose to disease relapses.
Genetic Predisposition
A familial aggregation study found a relative risk of 1.56 for GPA among first-degree relatives of patients with GPA, which is similar to the risk seen in rheumatoid arthritis [15]. Familial clustering has also been reported with other AAV syndromes [16, 17]. Several candidate genes have been investigated in several different cohorts of patients with AAV, and single nucleotide polymorphisms (SNPs) have been found in several genes coding for proteins involved in the immune response [18].
Variants most strongly and most reproducibly associated with AAV are found in the major histocompatibility complex (MHC) and PTPN22 genes [18]. Most genetic studies in AAV comprise only GPA cohorts, or the number of GPA patients included in the studied cohort is much larger than those with MPA and EGPA. Nevertheless, interesting genetic differences are emerging between the different syndromes. For instance, the HLA-DPB1*0401 variant represents a strong and reproducible genetic risk factor for GPA, but not for MPA or EGPA [19–21]. Interestingly, African-Americans with PR3-ANCA-associated vasculitis had a 36-fold higher likelihood to have the HLA-DRB1*15 genotype than community-based controls, and the HLA-DRB*1501 allele, which is of Caucasian rather than African descent, conveyed a 73.3-fold higher risk for PR3-ANCA-associated vasculitis [22].
In contrast, an increase in the PTPN22 620W allele, which has been associated with other autoantibody-associated autoimmune diseases and has thus been implicated in the regulation of B lymphocyte activity, was found to be associated with both GPA and MPA [23–25].
Cytotoxic T lymphocyte antigen 4 (CTLA4), expressed mostly on CD4-positive T lymphocytes, exerts an inhibitory function on T lymphocytes by binding to CD80 and CD86 on antigen-presenting cells. CTLA4 competes with the co-stimulatory molecule CD28, which exerts a stimulatory effect on T lymphocytes, for binding to CD80 and CD86. If T lymphocytes are activated via the T cell receptor and CD28, CTLA4 expression is increased, probably as a regulatory mechanism. Via these mechanisms CTLA4 is thought to maintain peripheral self-tolerance [26]. Elevated levels of CTLA4 have been found in GPA, undoubtedly a reflection of T lymphocyte activation [27]. CTLA4 is coded for by the CTLA4 gene, and CTLA4 polymorphisms, which may negatively affect expression or function of CTLA4, have been associated with autoimmune diseases including GPA [24, 28, 29].
Another gene of interest is PRTN3, which codes for PR3, the most prominent target antigen for ANCA in GPA. Eight SNPs have been identified in the PRTN3 promotor region and exons, but their function for PR3 expression is unclear. An increased frequency of one promotor polymorphism (A546G) was found among patients with GPA [30]. PR3 is not only stored in granules and released during neutrophil activation, but it is also expressed on the neutrophil membrane, where it may be engaged by PR3-ANCA (see below). The membrane expression of PR3 varies between individuals, remains constant over time, and appears genetically determined [31, 32]. One study found a link between membrane PR3 expression and HLA antigens [33]. Individuals with high membrane PR3 expression are significantly more frequent among patients with GPA than in the normal population, and patients with GPA and high membrane PR3 expression are at higher risk for relapses than those with low membrane PR3 expression [34, 35].
The major natural inhibitor for PR3 is alpha 1-antitrypsin, coded for by the AAT gene. Of the many polymorphisms of AAT, the Z and S alleles are responsible for the majority of alpha 1-antitrypsin deficiency cases. An increased frequency of heterozygosity for the Z and S alleles in association with GPA, but not MPA, has been reported by several groups in small studies and recently confirmed in larger studies [36, 37]. The clinical significance of this association is not entirely clear, but heterozygosity for the Z allele has been associated with a worse prognosis of the disease [38]. This may be because Z-allele carriers, but not S-allele carriers, have increased levels of pro-inflammatory polymers circulating in their blood [37]. Deposits of these polymers have been documented in kidney biopsy specimens from patients with active GPA, and these polymers can prime neutrophils and augment the activation of neutrophils by PR3-ANCA in vitro [37].
Unconfirmed or conflicting results have been reported for the association with either GPA or MPA and polymorphisms of other genes including those coding for the high-affinity soluble interleukin 2 receptor (IL-2RA), interleukin 10 (IL-10), leukocyte immunoglobulin-like receptor A2 (LILRA2) and CD226 (CD226), as well as for the Fc receptors FcRIIa and FcRIIIb [18].
Another, yet unclear, factor to be considered in the interpretation of genetic SNP association studies is the issue of copy number variation [39]. Copy number variations as a result of gene duplication, triplication, or exon shuffling may occur at a higher frequency in the genome than SNPs and may be more important for evolution [39]. The study of effects of copy number variations on the disease phenotype of complex autoimmune diseases including the AAV syndromes is just beginning.
Environmental Triggers and Infection
Environmental triggers for the onset of AAV in susceptible individuals remain unknown for most patients. A significant association between MPA with MPO-ANCA and exposure to silica has been reported [40].
The onset of AAV (predominantly MPA) has also been reported following exposure to a variety of therapeutic agents including propylthiouracil, hydralazine, and penicillamine [41, 42]. These agents have polyclonal B lymphocyte stimulatory properties, which may induce the production of ANCA. However, there are several features distinguishing drug-induced AAV from typical AAV. First, the ANCA response is often targeting several different antigens at the same time, whereas in typical AAV ANCA target either PR3 or MPO, but not both and not other additional antigens such as human neutrophil elastase or lactoferrin. Second, drug-induced AAV usually subsides after discontinuation of the offending agent. Thus, the cases of drug-induced AAV support the hypothesis of the pathogenic role of ANCA in the development of vasculitis (see below), but they do not explain the loss of tolerance to ANCA target antigens characterizing GPA and MPA.
Infections have been implicated as triggers as well as persistent drivers of various autoimmune diseases including AAV. A variety of different and often interrelated mechanisms by which infections trigger and perpetuate the disease in predisposed patients have been proposed. Many infectious agents have been reported to induce an ANCA response. In most instances these ANCA are directed against target antigens other than MPO or PR3. Furthermore, these infection-associated ANCA usually disappear once the infection resolves (reviewed in [2, 43, 44]). Thus, it seems that two conditions need to be met for infections to trigger AAV. First, tolerance to self-antigens needs to be broken, i.e., host conditions permissive for the development of autoimmunity need to allow for the ongoing production of antibodies directed against self-antigens (ANCA). Second, the persistent specific autoantibodies (ANCA) need to have pathogenic potential for the development of tissue injury characteristic of AAV.
One concept by which infections can elicit an autoimmune response in susceptible hosts is molecular mimicry [45]. Subsequent diversification of T and B lymphocyte responses (“epitope spreading”) may lead to reactivities with different epitopes on the same target molecule (intramolecular spreading) or even extend to other molecules (intermolecular spreading) [46, 47].
The only example of direct epitope mimicry leading to ANCA in patients with pauci-immune glomerulonephritis has been reported by Kain and co-workers [48]. They found ANCA targeting lysosomal membrane protein-2 (LAMP-2) in the majority of patients with pauci-immune focal necrotizing glomerulonephritis [48]. These ANCA recognized an epitope on LAMP-2 and cross-reacted with the homologous bacterial adhesin FimH [48]. Antibodies to LAMP-2 transferred to rats caused pauci-immune glomerulonephritis in the recipients [48]. Rats immunized with FimH developed antibodies to FimH that cross-reacted with human LAMP-2 and also developed pauci-immune glomerulonephritis [48]. The frequency of LAMP-2-specific ANCA has recently been confirmed in different patient cohorts from Europe [49], but not from the United States [50]. It also appears that the LAMP-2-specific ANCA disappear quickly after initiation of immunosuppressive therapy, and methods for their detection are not as robust as those used for the detection of PR3-ANCA and MPO-ANCA [49]. Consequently, the clinical relevance of this finding remains controversial.
An indirect mechanism of molecular mimicry leading to typical PR3-ANCA has been proposed by Pendergraft and colleagues [51]. According to this theory, idiotypic networks are instrumental in the formation of ANCA [52]. First, antibodies are formed against complementary peptides (antisense peptide sequence) to PR3 (cPR3) [51]. The cPR3 peptides, which may represent mimics of microbial peptide sequences, are the target of the primary immune response. Indeed, several bacterial peptide sequences including Staphylococcus aureus (S. aureus) sequences were found to have homologies with cPR3 [51]. True PR3-ANCA are subsequently the result of a secondary immune response mounted against the idiotype of these anti-cPR3 antibodies [51]. Pendergraft et al. were able to show that mice immunized with cPR3 developed antibodies against both cPR3 and PR3, and they found antibodies against cPR3 in 7 of 34 PR3-ANCA-positive patients [51]. However, the same group was unable to demonstrate a similar scenario for MPO-ANCA [53]. Furthermore, Tadema et al. could not confirm an increased frequency of anti-cPR3 antibodies in patients with GPA compared to healthy volunteers or MPO-ANCA-positive patients [54]. Interestingly, portions of the cPR3 sequence have homology with portions of the plasminogen sequence, and anti-plasminogen antibodies were detected in some patients with AAV, potentially contributing to the well-recognized increased risk for thromboembolic events [55, 56].
Direct or indirect epitope mimicry may explain how autoantibodies are formed, but not how they escape elimination by mechanisms that preserve immune tolerance to self-antigens. A variety of mechanisms by which infections promote the autoimmune response as well as the propensity for chronic relapses in AAV have recently been studied. Nearly two thirds of patients presenting with GPA are nasal carriers of S. aureus [57]. This is much higher than in the general population. S. aureus carriers are at higher risk for relapse of GPA than noncarriers, and treatment with trimethoprim–sulfamethoxazole resulted in a significant reduction of the relapse rate [57–59]. Multiple mechanisms may contribute to the increased risk of relapse conveyed by S. aureus.
S. aureus produces superantigens known to be powerful nonspecific (antigen-independent) T and B lymphocyte activators able to induce significant cell proliferation and cytokine release [60, 61]. Patients colonized with superantigen-producing strains of S. aureus are at higher risk for disease flares than those colonized with superantigen-negative strains [62]. Compared to healthy controls, patients with GPA were found to have an expansion of T lymphocytes expressing Vbeta segments specific for S. aureus superantigens [63]. However, a direct association between the presence of S. aureus producing superantigens and the expansion of T lymphocytes reactive to these superantigens in individual patients could not be confirmed [63].
S. aureus-derived superantigens and peptidoglycans as well as fungal beta-glucans can induce the expansion of IL-17-producing CD4-positive T cells, the so-called Th17 cells, in an IL-23-dependent manner [64–66]. Th17 cells, which are now recognized as central players in the development of autoimmunity, are highly potent inflammatory cells that initiate and maintain tissue inflammation by recruiting other inflammatory cells while generating a milieu that makes them resistant to control by T-regulatory (Treg) cells [67, 68]. IL-23 produced by antigen-presenting cells appears necessary to maintain Th17 cells at the site of inflammation [68]. Both elevated IL-23 and IL-17 levels have been found in patients with active disease and remained elevated despite treatment during clinical remission [69]. An increased frequency of Th17 cells responding to staphylococcal enterotoxin B was found in GPA patients in remission compared to healthy controls, regardless of ANCA status, whereas an increased frequency of PR3-responsive Th17 cells was restricted to PR3-ANCA-positive patients [70]. IL17 can also induce IL1-beta and TNF-alpha production and release by macrophages, and these cytokines prime neutrophils and monocytes, resulting in the expression of ANCA target antigens on their surface [71]. Chronic infections may thus set the stage for chronic ongoing inflammation and the loss of self-tolerance.
Necrotizing granulomatous inflammation consisting of monocytes, macrophages, neutrophils, T cells, B cells, and plasma cells, predominantly located in the respiratory tract, sets GPA clinically apart from MPA [1]. Structures resembling germinal centers have been documented within the granulomatous inflammatory tissue of GPA, and the tissue also stained positive for PR3 [72]. The immunoglobulin (VH) gene mutational patterns obtained from granulomatous nasal tissue of patients with GPA suggest that the selection and maturation of PR3-ANCA-producing B lymphocytes may start within the granulomatous lesions [72].
Cytosine–phosphate–guanine (CPG) motifs are pathogen-associated molecular patterns (PAMPs) that are recognized by the pattern recognition receptor Toll-like receptor 9 (TLR9). Unmethylated CPG oligodeoxynucleotides (CpG-ODN) are potent immune stimulants. B lymphocytes isolated from patients with AAV with active disease as well as during remission can be induced to produce ANCA when exposed to CpG-ODN and IL-2 [73, 74]. Significantly, more PR3-ANCA patients than MPO-ANCA patients produced ANCA in vitro in response to CpG exposure [74]. These observations provide another link between S. aureus infection, granulomatous inflammation of the respiratory tract, and the higher relapse rate of patients with PR3-ANCA or GPA compared to patients with MPO-ANCA or MPA.
Neutrophils are key players of the innate immune defense against microorganisms. An additional antimicrobial defense mechanism of neutrophils has been described recently—a unique type of cell death distinct from apoptosis and necrosis that is associated with the formation and release of neutrophil extracellular traps (NETs) [75]. NETs are extracellular structures containing chromatin and granule proteins (including ANCA target antigens) in which invading microbes are trapped and killed. The formation of NETs depends on reactive oxygen species generated by NADPH oxidase [76]. ANCA, which are known to induce a respiratory burst in primed neutrophils, can also induce the formation of NETs, and ANCA antigens bound to NETs are accessible to ANCA [77]. NETs containing PR3 and MPO are detectable in the kidneys of patients with AAV in the absence of infection. Interestingly, S. aureus can rapidly and strongly induce NETs formation even without causing neutrophil death [78]. Chromatin-immunoglobulin complexes are thought to lead to the loss of tolerance and autoantibody (ANCA) production in a TLR9-dependent manner [79]. Taken together, these observations suggest that ANCA may induce a vicious cycle of self-perpetuated NET formation and more ANCA production, particularly in the presence of S. aureus infection. This hypothesis is further supported by observations of increased TLR9 expression by monocytes of patients with GPA who were S. aureus nasal carriers [80].
Taken together, this evidence suggests that S. aureus and possibly other microbial organisms contribute to the pathogenesis of GPA and MPA by creating a permissive inflammatory environment by promoting T and B lymphocyte activity, which in turn can initiate and maintain ANCA production in predisposed patients. Both the chronic inflammatory milieu with ongoing cytokine release and the loss of tolerance to the ANCA target antigen appear to be required for the cascade that results in necrotizing small vessel vasculitis.
The Pathogenic Role of Lymphocytes
ANCA are high-affinity class-switched antibodies [81]. This implies that ANCA production is dependent on T lymphocyte help, that autoreactive T lymphocytes are present, and that there is insufficient counter-regulation by Tregs. Indeed, T lymphocyte abnormalities have long been suspected to be the main reason for the chronic relapsing nature of GPA [82]. Patients with GPA in remission have an increased percentage of circulating CD4-positive effector memory T lymphocytes as well as a functional defect of circulating CD4-positive CD25-positive Tregs [83–85]. Moreover, patients with reduced numbers of Tregs required more prolonged treatment to achieve remission, and their relapse rates were higher [85].
The role of B lymphocytes in the pathogenesis of AAV appears more clearly defined than that of T lymphocytes. First, B lymphocytes have been found in affected tissues including kidneys and most significantly in granulomatous lesions of the respiratory tract, where they are located in close proximity to numerous PR3-positive cells and where the selection and maturation into PR3-ANCA-producing B lymphocytes may occur [72]. Second, antigen-specific B lymphocytes are the progenitor cells of short-lived plasma cells thought to be the source of autoantibodies including ANCA [86, 87]. Third, the proportion of circulating activated B lymphocytes is increased in patients with GPA compared to healthy controls, and it is higher in patients with active disease versus those in remission and in patients with generalized disease versus those with limited disease [82]. Most importantly, the beneficial therapeutic effects of cyclophosphamide (CYC) in GPA have been attributed to its effect on B lymphocytes, and the pathogenic role of B lymphocytes explains why rituximab, which targets these cells selectively, is so successful at controlling the disease activity of GPA [88, 89].
The Pathogenic Role of ANCA
What do ANCA do once they are formed? Clinical observations and a large body of experimental data support a pathogenic role of ANCA for the development of small vessel vasculitis. The small vessel injury of AAV appears to be caused by activated leukocytes.
The pro-inflammatory pathogenic effects of ANCA are all contingent on their interactions with their target antigens expressed on the surface of primed neutrophils and monocytes. When primed with inflammatory cytokines such as tumor necrosis factor (TNF)-α[alpha] or microbial products in vitro, leukocytes express proteinase 3 (PR3) and myeloperoxidase (MPO) on their surface [90, 91]. PR3-ANCA and MPO-ANCA can activate primed neutrophils and monocytes by binding directly to their antigens expressed on the surface or by Fc-receptor engagement; these interactions initiate signal transduction cascades via multiple pathways that are similar in neutrophils and monocytes [92–96].
The activation of neutrophils and monocytes by ANCA has several pro-inflammatory effects that in aggregate cause tissue injury. Fully activated neutrophils degranulate and release toxic proteases and enzymes including elastase, PR3, MPO, and others [90]. ANCA also induce a respiratory burst resulting in the release of oxygen radical species [90, 97]. Furthermore, ANCA induce the expression of cell adhesion molecules on neutrophils and endothelial cells leading to an increased adhesion of neutrophils to endothelial cells [98–102]. Moreover, the binding of ANCA to primed leukocytes induces the production and release of chemotactic cytokines including IL-1, MCP-1, and IL-8 [103–106]. These cytokines attract more neutrophils and monocytes to the site of inflammation. Thus, when the ANCA-induced cytokine release occurs at the endothelial interface, the normal chemotactic gradient that draws neutrophils out of the vasculature into the tissues is lost. This causes further accumulation of fully activated neutrophils in the vessel wall, where they cause more injury.
The ANCA target antigens released from activated or dying neutrophils can also directly bind to endothelial cells [107]. This may result in apoptosis of endothelial cells and to localized immune-complex formation with circulating ANCA [108]. Low levels of localized immune-complex deposition, which has been documented in early vasculitic skin lesions as well as renal lesions, can in turn induce localized complement activation [109, 110].
Capillaritis of the lung in AAV is characterized by fibrinoid necrosis and leukocytoclasis (Fig. 10.2). In vitro studies have suggested that ANCA modify the clearance of apoptotic cells. Opsonization of pre-apoptotic cells by ANCA is associated with an increased production of inflammatory cytokines by phagocytozing macrophages [111]. Moreover, pre-apoptotic cells have a decreased cell surface expression of phosphatidylserine (the recognition signal for macrophages) in the presence of ANCA [112]. Consequently, in the presence of ANCA, the noninflammatory clearance of apoptotic cells by macrophages may be perturbed in favor of inflammation and necrosis.
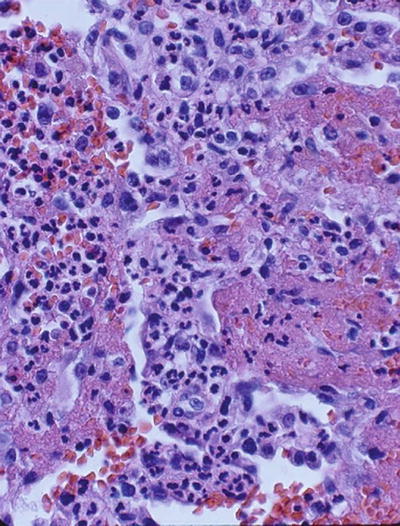
Fig. 10.2
Lung biopsy showing characteristic fibrinoid necrosis, hemorrhage, and leukocytoclasis (H & E, ×40)
Several in vivo animal models support the pathogenic role of ANCA. These models are based on the transfer of antibodies generated against ANCA target antigens into healthy recipient animals. The transfer of anti-MPO IgG or splenocytes obtained from MPO-knockout mice, which were immunized with murine MPO, into Rag 2 knock-out mice (lacking mature T- and B lymphocytes) as well as into wild-type mice resulted in pauci-immune crescentic necrotizing glomerulonephritis similar to that found in humans [113]. A direct augmenting effect of MPO-ANCA on neutrophil–endothelial interactions causing microvascular injury was documented in a rat anti-MPO antibody transfer model [114]. Murine anti-PR3 antibodies generated in a similar fashion only caused an increased inflammatory response at the site of tissue injury, but not a vasculitic phenotype, when transferred into wild-type mice [115].
Even though patients with active AAV have normal serum complement levels and lack significant immune-complex deposits in affected tissues, low-grade localized immune-complex formation and complement activation may play a role. It has recently been recognized that activation of the alternative complement pathway by ANCA may represent an important amplification loop of inflammation that contributes to renal (and other tissue) injury in AAV. In the murine anti-MPO antibody transfer model, the development of necrotizing glomerulonephritis is dependent on the activation of the alternative complement pathway, and the development of lesions can be prevented and treated with an antibody that inhibited complement factor 5 (C5) activation [116, 117]. Mice lacking the receptor for activated C5 on neutrophils also do not develop the renal lesions [118]. In vitro studies showed that supernatants from ANCA-activated neutrophils can cause the production of C5a in normal serum, C5a receptor-dependent priming of normal neutrophils, and the increased neutrophil membrane expression of PR3. Even though the renal lesions in humans are called “pauci-immune,” the components of the alternative complement pathway can be detected in patients with AAV, but not normal controls or patients with minimal change disease [119].
Despite all this evidence, proof that ANCA alone can cause disease in humans has remained elusive. One case study in which an infant was born to a mother with active MPA is often quoted as such evidence [120]. The infant developed a pulmonary–renal syndrome 48 h after delivery and was found to have serum MPO-ANCA titers similar to the mother [120]. The child was treated with glucocorticoids and plasma exchange and recovered. However, this observation is countered by another report of a case where MPO-ANCA were also transferred from the mother to the newborn, but the newborn remained perfectly healthy despite persistence of the transferred MPO-ANCA in the newborn for several weeks [121]. The two contrasting case studies are consistent with observations made in large cohort studies. The development of severe vasculitic disease manifestations and severe flares usually do not occur in the absence of ANCA, but not all patients with persistent ANCA inevitably suffer such flares [6, 8, 57, 122].
The multiple clinical and experimental observations are best reconciled by the hypothesis that ANCA alone do not cause the disease, but in an inflammatory environment, they are conditional for the progression to systemic small vessel vasculitis. Therapeutic interventions that prevent the production of ANCA, remove them from the circulation, and interfere with their binding to target antigens or with downstream effects of this interaction all have a good scientific rationale. The clinical challenge is to achieve this in a targeted fashion with minimal adverse effects.
Management of Patients with ANCA-Associated Vasculitis
Diagnostic Approach
The patient’s symptomatology at presentation is determined by the type, extent, and location of the inflammatory lesions. The initial diagnostic approach needs to be adapted according to the initial clinical presentation. However, AAV in all its flavors is a systemic disorder, and some disease manifestations may not cause overt symptoms. Therefore, every patient suspected of having a small vessel vasculitis should undergo initial testing that includes a complete blood count with differential, metabolic panel with serum creatinine and transaminases, urinalysis and microscopy, erythrocyte sedimentation rate, C-reactive protein, and ANCA determinations as well as a chest imaging study to look for clinically silent lung involvement. Tests for other autoimmune disorders (e.g., ANA, anti-GBM, anti-double-stranded DNA antibodies, antiphospholipid antibodies, and cryoglobulins) should also be part of the initial diagnostic evaluation.
The diagnostic value of ANCA testing is widely accepted. Yet, ANCA testing is not standardized, and differences in analytical sensitivity and specificity exist between different assays [123–126]. In AAV, ANCA causing a cytoplasmic immunofluorescence pattern (C-ANCA) on ethanol-fixed neutrophils are caused by antibodies reacting with PR3. A perinuclear immunofluorescence pattern (P-ANCA) can be caused by antibodies reacting with a variety of neutrophil antigens. However, only those that react with MPO are important in AAV [2]. In order to assure diagnostic accuracy of ANCA testing, both a target antigen-specific test (PR3-ANCA or MPO-ANCA) and immunofluorescence should be performed [127]. The two test results should confirm each other. Only the PR3-ANCA with C-ANCA combination and MPO-ANCA with P-ANCA are sensitive and relatively specific for AAV [124, 127]. In addition, the positive and negative predictive value of an ANCA test result depends on the pretest probability of the disease in the patient tested [124, 128–130]. PR3-ANCA are the predominant ANCA type in GPA, and only 5–10 % of patients with GPA have MPO-ANCA. It is important to note that about a quarter of patients with limited GPA have no detectable ANCA [6]. Yet, such patients may subsequently seroconvert and develop severe disease. Recently, the diagnostic interpretation of ANCA test results has been further complicated by emerging pathology associated with the widespread exposure to levamisole among habitual cocaine users [131, 132].
More invasive diagnostic tests may be necessary to fully assess the disease extent and severity in individual patients. The presence of respiratory symptoms or chest roentgenographic abnormalities should prompt complete pulmonary function testing including inspiratory and expiratory flow volume loop determinations. Respiratory symptoms that cannot be explained satisfactorily based on physical examination and roentgenographic findings, roentgenographic abnormalities suggesting the possibility of alveolar hemorrhage, and abnormalities in the shape of the flow volume loops, all represent indications for bronchoscopic evaluation to establish or rule out endobronchial disease of GPA or alveolar hemorrhage in GPA and MPA [133].
A renal biopsy may be necessary to establish a diagnosis, and it may provide prognostic information (Fig. 10.3). Renal outcome at 1 year can be estimated using the GFR at baseline in combination with histologic information [134]. Based on renal biopsies obtained during clinical trials performed in Europe, an international working group of renal pathologists has recently proposed a classification system for glomerular lesions that was validated to be of prognostic value for 1- and 5-year renal outcomes (Fig. 10.4) [135]. Tubular atrophy, interstitial fibrosis, and presence of intraepithelial T lymphocytes on a biopsy specimen were also found to be predictors of estimated glomerular filtration rate at 1 year, but only tubular atrophy remained an independent predictor of renal function at 2 years [136]. Despite these emerging data, the ultimate renal prognosis of an individual patient cannot be estimated conclusively based on renal biopsy findings, and treatment decisions should not be based on biopsy findings alone.
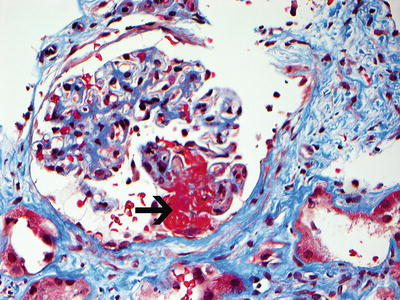
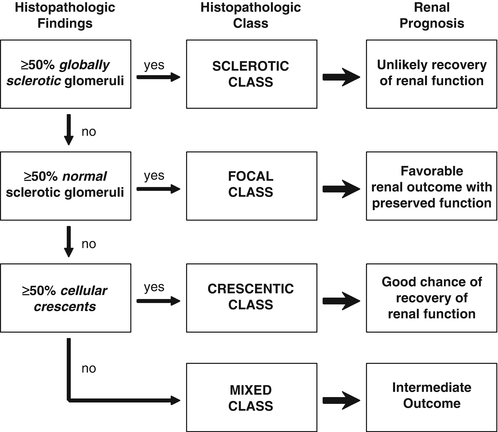
Fig. 10.3
Necrotizing glomerulonephritis with a glomerulus showing segmental fibrinoid necrosis (black arrow points to necrosis). Note disruption of the glomerular tuft with extension of necrosis into the Bowman’s space (Trichrome stain, ×40). Contrast with thrombotic microangiopathy in which the thrombus is present within the capillary loops (Courtesy of Dr. Sanjeev Sethi, Mayo Clinic)
Fig. 10.4
Histologic categories and renal outcome in ANCA-associated vasculitis. Phenotypical order of categories (focal, crescentic, mixed, and sclerotic) corresponds to the order of severity of renal function impairment. Modified from Journal Am Soc Nephrol 2010; 21(10):1628–1636. With kind permission from the American Society of Nephrology
Prognostic Factors
Several studies have shown that the following factors negatively impact survival in patients with AAV and renal involvement: age at diagnosis, glomerular filtration rate at diagnosis, end-stage renal disease (ESRD) at diagnosis, and PR3-ANCA [137–140]. Consequently, to avoid irreversible damage and death, swift implementation of definitive immunosuppressive therapy is crucial.
Remission Induction Therapy for Generalized or Severe AAV
Our current treatment approach to GPA and MPA is based on the results of several randomized controlled trials (Table 10.1). Patients with GPA are categorized based on disease activity and extent into having non-severe (limited) or severe disease, whereas most disease manifestations of MPA lead to the categorization of severe disease. Severe disease refers to disease manifestations that are either life threatening or have the potential to cause irreversible damage in affected organs. Severe disease manifestations include alveolar hemorrhage, glomerulonephritis, neuropathy, scleritis, and heart and gastrointestinal involvement and are usually caused by capillaritis or small vessel vasculitis. In contrast, the non-severe disease manifestations of GPA are usually caused by necrotizing granulomatous inflammation. Sometimes the granulomatous inflammatory disease burden is so extensive or is localized such that it can be life threatening. Such patients are also considered to have severe disease for treatment purposes.
Table 10.1
Randomized controlled trials informing the management of ANCA-associated vasculitis
Treatment investigated | Trial design | Patients randomized (randomization schedule) | Main conclusion | Trial moniker | Reference |
|---|---|---|---|---|---|
TMP/SMX for remission maintenance in GPA | Randomized, placebo controlled | 81 (1:1) | TMP/SMX reduces incidence of relapses in WG | N/A | [57] |
AZA versus CYC for remission maintenance in severe AAV | Randomized, controlled, open label | 144 (1:1) | AZA is as effective for remission maintenance as continuation of CYC in severe AAV | CYCAZAREM | [143] |
MTX versus CYC for remission induction in early systemic AAV | Randomized, controlled, open label | 100 (1:1) | MTX is not inferior to CYC in early systemic AAV | NORAM | [142] |
PLEX versus intravenous methylprednisolone for AAV with severe renal disease | Randomized, controlled, open label | 137 (1:1) | PLEX improves renal survival of methylprednisolone in AAV with severe renal disease | MEPEX | [153] |
AZA versus MTX for remission maintenance in severe AAV following induction with CYC | Randomized, controlled, open label | 126 (1:1) | There is no difference in efficacy (relapse rate) or adverse events rate between AZA and MTX | WGENT | [159] |
Etanercept versus placebo added to standard therapy for limited and severe GPA | Randomized, double blind, placebo controlled | 180 (1:1) | Etanercept has no benefit in addition to standard therapy | WGET | [144] |
Pulse CYC versus oral daily CYC for remission induction in severe AAV | Randomized, controlled, open label | 149 (1:1) | Pulse CYC is as effective as oral daily CYC for severe AAV | CYCLOPS | [146] |
MMF versus AZA for remission maintenance following induction with CYC | Randomized, controlled, open label | 156 (1:1) | MMF is less effective than AZA for remission maintenance | IMPROVE | [160] |
RTX versus oral daily CYC followed by AZA for remission induction in severe AAV | Randomized, double blind, double placebo controlled | 197 (1:1) | RTX is not inferior to CYC for remission induction in severe AAV and RTX is superior to CYC for relapsing patients | RAVE | |
RTX versus pulse CYC followed by AZA for remission induction in severe AAV with renal disease | Randomized, controlled, open label
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




