Fig. 19.1
Collagenofibrotic glomerulopathy (PAS stain). From Ponticelli and Glassock [1]; by permission of Oxford University Press
The mechanisms responsible for the deposition of type III collagen in glomeruli are uncertain [1–5]. Elevated circulating levels of procollagen type III peptides suggest a systemic disease; but, no similar deposits have been detected in other organs. Hepatic fibrosis can occasionally be observed. On the other hand, mesangial cells and podocyte might acquire the capacity to synthesize and to secrete type III collagen under the influence of an abnormal cytokine milieu (such as excess Interleukin 4) or a genetic mutation [5].
The disorder usually slowly progresses to end-stage renal disease (ESRD) often after many years. The renal survival rate is about 50 % at 10 years from diagnosis. No specific treatment is available, but control of hypertension and use of angiotensin inhibition may delay ESRD [1–5]. A role for steroid treatment has been suggested by anecdotal improvement in proteinuria and procollagen type III levels. Experience with renal transplantation is limited, but levels of procollagen type III peptides increase rather than decrease posttransplantation. No recurrent disease has yet been reported, but follow-up times are short (<5 years) [1–5].
Lipoprotein Glomerulopathy
This is a rare disorder identified by thrombotic-like deposits of lipoproteins in glomeruli [1, 6–9]. In many cases a hereditary dispostion is noted, linked to the ApoE gene on Chromosome 19. A familial distribution is found in about one-third of a cases, but the specific mode of inheritance is not fully understood. The clinical findings at presentation are nonspecific and consist of proteinuria (often in the nephrotic range), hypertension, and impaired renal function. Almost any age or gender can be affected, but patients are usually between the age of 20 and 40 years at the time of discovery [1, 6–9]. Hyperlipidemia (hypertriglyceridemia—type II phenotype) is often present and elevated plasma apoE is almost always present [1, 6–9].
The diagnosis is made by renal biopsy. By light microscopy the glomerular capillaries are distended and filled with a hematoxylin and eosin pale-staining substance positive for lipid (Oil Red O stain) [6–9] (Fig. 19.2). These deposits contain apoE, apoB, and apoA by immunofluorescence microscopy, and Ig are absent. Electron microscopy shows deposits of various sizes forming lamellated structures. No fibrils are present [6–9].
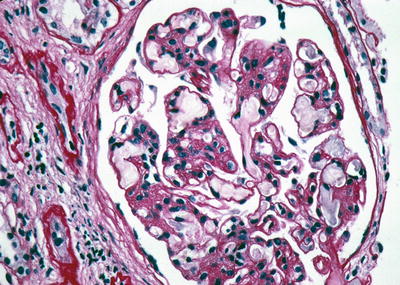
Fig. 19.2
Lipoprotein glomerulopathy (PAS stain). From Ponticelli and Glassock [1]; by permission of Oxford University Press
The disease is likely caused by heterogenous mutations in the apoE gene (e.g., apoE Sendai, Tokyo, Maebashi) resulting in an instability of the molecule and its aggregation in glomerular capillaries [10, 11]. ApoE is also synthesized by glomerular cells and can regulate glomerular mesangial function in an autocrine fashion, including that of Fc receptors [10]. Dysfunction of the latter may participate in the accumulation of lipoprotein deposits in the mesangial and endothelial cells by altering uptake and clearance.
The disease is usually slowly progressive, but at highly variable rates. Treatment with steroids or immunosuppressive agents is ineffective. The best treatment is with fibric-acid derivatives (fibrates) which can both slow progression and cause the disappearance of the lipoprotein deposits in addition to improving the hyperlipidemia [1, 6–9, 12]. Treatment with LDL apheresis and nicotinic acid derivatives (niceritrol) can also be beneficial [13, 14]. The disease can recur in renal transplants attesting to its systemic nature [1].
C1q Nephropathy
This is an uncommon, but not rare, disorder that can only be diagnosed with certainty by immunofluorescence microscopy (like IgA nephropathy) [1, 15–19]. It accounts for <3 % of all forms of primary glomerular disease causing nephrotic syndrome in adults and children [16–19]. Although it is more common in African-Americans, it has been described in many ancestral groups. Rarely familial cases have been described. Males predominate (about 2:1) and both adults and children can be affected [16–19]. The clinical features are nonspecific. Proteinuria (often in the nephrotic range), hypertension, and hematuria are common. Acute renal failure (often in association with gross hematuria) and progressive renal failure can develop [20, 21]. Rapidly progressive glomerulonephritis (with superimposed crescents) has been observed [8]. Systemic features suggesting an autoimmune disease are uniformly absent. Serum complement component values are typically normal, but rarely an acute nephritic syndrome with hypocomplementemia can develop [22]. Antinuclear antibodies and anti-C1q autoantibodies are not found [1, 16–19].
The diagnosis is exclusively made by renal biopsy and immunofluorescence microscopy [1, 16–19, 23, 24]. By light microscopy a variety of findings are present, including minimal change disease (MCD), mesangial proliferative glomerulonephritis (MesPGN), and focal and segmental glomerulosclerosis (FSGS). The latter is the most common lesion observed and can frequently be of the “collapsing variant.” The association with polymorphisms in MYH9 gene may explain the frequent association of C1q nephropathy and collapsing FSGS in African-Americans [25]. The characteristic findings are revealed in immunofluorescence microscopy [16–19]. Intense mesangial deposition of C1q accompanied by C3 in an amorphous or granular pattern is characteristic. Polyclonal IgG and to a lesser extent IgM is also found in 85–95 % of cases. This pattern resembles that seen in lupus nephritis, but no clinical or laboratory features of SLE are present (by definition). Electron microscopy shows mesangial electron-dense deposits, but tubuloreticular inclusions are rarely found.
The pathogenesis of this disorder is obscure [1, 16–19]. Certain features suggest in situ or circulating deposition of immune complexes. The antigen is unknown.
The clinical manifestation of renal disease tends to persist, but spontaneous remissions have been reported [26], especially following a bout of acute renal failure and gross hematuria. Progressive renal disease is the rule and the 3 year renal survival is about 80 % [1, 16–19]. Collapsing FSGS and persistent marked proteinuria portend a poorer prognosis [16–19].
Treatment is quite uncertain. Partial or complete remission of nephrotic syndrome can follow steroid treatment in about 10–20 % of cases, which is probably higher than a spontaneous remission rate [1, 16–19, 21]. No randomized controlled trials of treatment have been conducted. Patients with MCD probably respond better to steroid therapy than those with FSGS. The effect of cytotoxic immunosuppressive drugs and calcineurin inhibitors is largely unknown. Preliminary anecdotes suggest a possible role for rituximab therapy [27]. Recurrent disease in renal transplants has not yet been observed, but C1q deposition can be observed in renal transplant biopsies, irrespective of the original disease [28].
Idiopathic Nodular Glomerulosclerosis
The development of “nodular” lesions in an intercapillary (mesangial) distribution is a common finding in advanced diabetic nephropathy (the Kimmelstiel–Wilson lesion) and in some forms of membranoproliferative glomerulonephritis and monoclonal light-chain deposition diseases as well [1, 29–32]. When diseases known to provoke this pattern of glomerular histology are absent, one can designate the disorder as “idiopathic” nodular glomerulosclerosis (ING) [29–32].
It is a rather uncommon lesion and tends to be observed in older adults (typically >65 years of age), especially women, and in the presence of a history of heavy smoking [29–32]. It has not been reported in children. All cases are sporadic. Blood sugar and hemoglobin A1c levels are normal (by definition), and no monoclonal gammopathies should be present (serum free light chain kappa/lambda ratios should be normal). Serum complement component levels are normal. Dyslipidemia, hypertension, proteinuria (including nephrotic syndrome), and atherosclerosis are common [1, 29–32].
By light microscopy the lesion is characterized by diffuse and nodular mesangial sclerosis and thickening of the glomerular basement membrane (GBM) (Fig. 19.3) [29–32]. Severe arteriolar sclerosing lesions are also seen. Amyloid deposition is absent. Immunofluorescence microscopy may show diffuse linear albumin deposits along the GBM but Ig and C3 are absent. The electron microscopic features are nonspecific. No electron-dense or fibrillary deposits are present. The GBM is thickened.
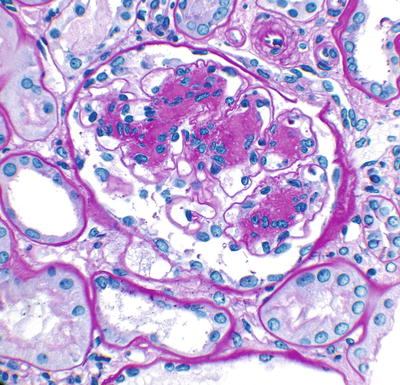
Fig. 19.3
Idiopathic nodular glomerulosclerosis (PAS stain). From Ponticelli and Glassock [1]; by permission of Oxford University Press
The pathogenesis of ING is unknown, but many features suggest chronic endothelial injury and repair, perhaps connected to smoking and intermittent renal hypoxia [28–32].
Most, but not all, patients progress to ESRD. Due to late discovery the median time from diagnosis to ESRD is about 2 years [1, 29–32]. Other than stopping smoking and control of dyslipidemia and hypertension (preferably with angiotensin-II inhibition), there is no known effective therapy for this condition. A recurrence in a renal allograft has been recently reported [33].
Fibrillary and Immunotactoid Glomerulonephritis
These two clinical-pathologic entities are often discussed together, even if they may have different underlying mechanisms [1, 34–39]. They share in common the presence of congo red negative (non-amyloid) deposits of immuno-globulins (Ig) that acquire a fibrillary (organized) substructure. They are often also called glomerulonephritis with organized monoclonal immunoglobulin deposits (GOMID) when the deposits of Ig are monoclonal in nature. The main distinctions between fibrillary (FGN) and immunotactoid (ITGN) glomerulonephritis are that the fibrils are smaller in FGN (12–24 nM in diameter) compared to ITGN (>30 nM in diameter and microtubular in appearance with parallel arrays) and that ITGN is much more commonly associated with a monoclonal gammopathy [34–39].
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


