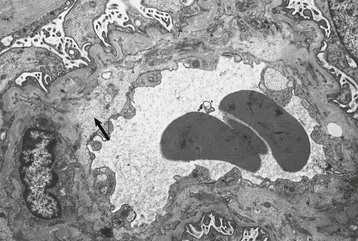
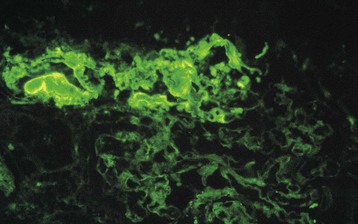
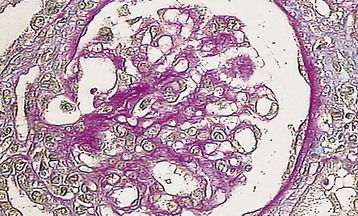
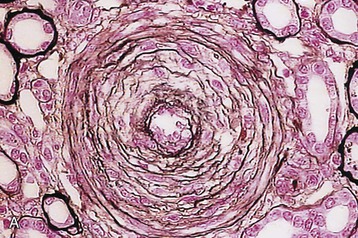
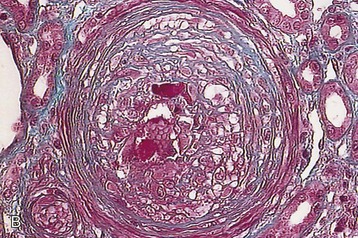
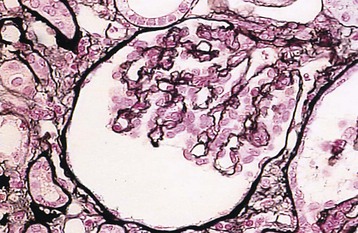
Marina Noris, Piero L. Ruggenenti, Giuseppe Remuzzi Thrombotic microangiopathy (TMA) is a lesion of arteriolar and capillary vessel wall thickening with intraluminal platelet thrombosis and a partial or complete obstruction of the vessel lumina. Laboratory features of thrombocytopenia and microangiopathic hemolytic anemia are almost invariably present in patients with TMA lesions and reflect the consumption and disruption of platelets and erythrocytes in the microvasculature. Depending on whether renal or brain lesions prevail, two pathologically indistinguishable but somehow clinically different entities have been described: the hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP). Because HUS can involve extrarenal manifestations and TTP may be associated with severe renal disease, the two can be difficult to distinguish on clinical grounds only.1 Newly identified pathophysiologic mechanisms, however, have allowed for the differentiation of the two syndromes on a molecular basis (Table 29-1). These new data also show, that, for example, pregnancy-associated HUS is genetically determined (Table 29-1) with pregnancy or transplantation acting as a trigger of TMA in a genetically predisposed individual. Table 29-1 Classification of hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) according to clinical presentation and underlying etiology. HELLP, Hemolytic anemia, elevated liver enzymes, and low platelet count; HSC, hematopoietic stem cell; SLE, systemic lupus erythematosus; APS, antiphospholipid syndrome. For all other abbreviations, refer to Table 29-3. The term hemolytic uremic syndrome was introduced in 1955 by Gasser and coworkers2 in their description of an acute fatal syndrome in children characterized by hemolytic anemia, thrombocytopenia, and severe renal failure. HUS occurs most frequently in children younger than 5 years, in whom the incidence is 5 to 6 children per 100,000 children population per year, compared with an overall incidence of 0.5 to 1/100,000/yr. Most cases (>90% of pediatric cases) are associated with infection by Shiga-like toxin (Stx)–producing Escherichia coli (STEC). STEC-HUS occurs primarily in children, except in epidemics, when it may occur in patients with a wider age range. For example, from May to July 2011, several European countries, particularly northern Germany, experienced one of the largest STEC-HUS outbreaks ever reported, with 3816 patients who had E. coli O104:H4 infection, with 845 cases. Almost 90% of affected patients were adults and, compared to previous STEC epidemics, there was a higher prevalence of affected young and middle-aged women.3 Streptococcus pneumoniae causes a distinctive form of HUS, accounting for 40% of cases not associated with Stx-producing bacteria.4 Approximately 10% of HUS cases are classified as “atypical,” caused neither by Stx-producing bacteria (STEC or Shigella dysenteriae) nor by Streptococcus.5 Atypical HUS is significantly less common than STEC-HUS, with an incidence of 0.5 to 2 per 1 million population per year.4 It occurs at any age. Patients with atypical HUS have a poor outcome; 50% progress to end-stage renal disease (ESRD), and 25% may die in the acute phase.4,6 Neurologic symptoms and fever can occur in 30% of patients. Pulmonary, cardiac, and gastrointestinal manifestations can also occur.4,6 Thrombotic thrombocytopenic purpura was first described in 1925 by Moschcowitz7 in a 16-year-old female patient with a fulminant febrile attack, hemolytic anemia, bleeding, renal failure, and neurologic involvement. Pathologic changes were characterized by widespread hyaline thrombosis of small vessels. TTP is a rare disease, with an incidence of approximately 2 to 4 cases per 1 million persons per year. TTP can affect any age group. TTP classically presents with the pentad of thrombocytopenia, microangiopathic hemolytic anemia, fever, and neurologic and renal dysfunction.7 Neurologic symptoms can be seen in more than 90% of patients over the course of the disease. Central nervous system (CNS) involvement mainly represents thrombo-occlusive disease of the gray matter, but can also include headache, cranial nerve palsies, confusion, stupor, and coma. Up to half of patients who present with neurologic involvement may be left with sequelae. Chronic kidney disease (CKD) may occur. One group has reported 25% of patients with creatinine clearance less than 40 ml/min receiving long-term follow-up. Cardiac involvement may be common in TTP patients.8 Laboratory features of thrombocytopenia and microangiopathic hemolytic anemia are almost invariably present in patients with TMA lesions and reflect consumption and disruption of platelets and erythrocytes in the microvasculature.5 Hemoglobin levels are low, less than 10 g/dl in more than 90% of patients. Reticulocyte counts are uniformly elevated. The peripheral smear reveals increased schistocyte numbers, with polychromasia and often nucleated red blood cells (RBCs) (Fig. 29-1). Detection of fragmented erythrocytes is crucial to confirm the microangiopathic nature of the hemolytic anemia, provided that heart valvular disease and other anatomic arterial abnormalities that may cause erythrocyte fragmentation are excluded. Other indicators of intravascular hemolysis include elevated lactate dehydrogenase (LDH), increased indirect bilirubin, and low haptoglobin level.5 The Coombs test is negative. Moderate leukocytosis may accompany the hemolytic anemia. Thrombocytopenia is uniformly present in HUS and TTP. It may be severe but is usually less so in patients with predominant renal involvement.9 The presence of giant platelets in the peripheral smear or reduced platelet survival time (or both) is consistent with peripheral consumption. In children with STEC-HUS, the duration of thrombocytopenia is variable and does not correlate with the course of renal disease. Bone marrow biopsy specimens usually show erythroid hyperplasia and an increased number of megakaryocytes. Prothrombin time, partial thromboplastin time, fibrinogen level, and coagulation factors are normal, thus differentiating HUS and TTP from disseminated intravascular coagulation (DIC). Mild fibrinolysis with minimal elevation in fibrin degradation products, however, may be observed. Evidence of renal involvement is present in all patients with HUS (by definition) and in about 25% of patients with TTP.1,10 Microscopic hematuria and subnephrotic proteinuria are the most consistent findings. STEC-HUS in 90% of patients is preceded by diarrhea, often bloody. The diagnostic histologic lesions of TMA consist of widening of the subendothelial space and microvascular thrombosis. Electron microscopy best identifies the characteristic lesions of swelling and detachment of the endothelial cells from the glomerular basement membrane and the accumulation of fluffy material in the subendothelium, intraluminal platelet thrombi, and partial or complete obstruction of vessel lumina11 (Figs. 29-2 and 29-3). These lesions are similar to those seen in other renal diseases, such as scleroderma, malignant nephrosclerosis, chronic transplant rejection, and calcineurin inhibitor nephrotoxicity. In HUS, microthrombi are present primarily in the kidneys; in TTP, they mainly involve the brain. In pediatric patients, particularly those younger than 2, and in those with STEC-HUS, the glomerular injury is predominant11 (Figs. 29-4 and 29-5). Thrombi and infiltration by leukocytes are common in the early phases of HUS and usually resolve after 2 to 3 weeks. Patchy cortical necrosis may be present in severe cases; crescent formation is uncommon. In idiopathic and familial forms and in adults, the injury mostly involves arteries and arterioles, with thrombosis and intimal thickening (Fig. 29-6; see also Fig. 29-3), and secondary glomerular ischemia and retraction of the glomerular tuft (Fig. 29-7). Focal segmental glomerulosclerosis may be a long-term sequela of acute cases of HUS and is usually seen in children with long-lasting hypertension and progressive deterioration in renal function. The typical pathologic changes of TTP are the thrombi that occlude capillaries and arterioles in many organs and tissues. These thrombi consist of fibrin and platelets, and their distribution is widespread. Thrombi are most frequently detected in kidneys, pancreas, heart, adrenals, and brain. Compared with HUS, pathologic changes of TTP are more extensively distributed, probably reflecting the more systemic nature of the disease. Shiga toxin–producing E. coli–associated HUS may follow gastrointestinal (GI) infection by certain strains of E. coli or S. dysenteriae that produce powerful exotoxins (Shiga toxins, Stx).12 Most patients present with bloody diarrhea that may still be active or that may resolve at presentation of HUS. Several strains of E. coli were found to produce Stx (STEC), mostly the serotype O157:H7, but also other serotypes, such as O111:H8, O103:H2, O123, O26, O145, and the O104:H4 strain of the recent German outbreak,3 isolated from patients with diarrhea. After food (beef, vegetables, fruit) or water contaminated by STEC or S. dysenteriae is ingested, the toxin is released into the gut and may cause watery or most often bloody diarrhea because of a direct effect on the intestinal mucosa. Stx-producing E. coli closely adhere to the epithelial cells of the GI mucosa, causing destruction of brush border villi. Shiga toxins are transported to the intracellular space of polarized GI cells via transcellular pathways and then translocated into the circulation. Circulating human blood cells (e.g., erythrocytes, platelets, monocytes) express Stx receptors on their surface and could serve as Stx carriers from the intestine to the kidney and other target organs. In the kidney, Shiga toxins bind mainly to glomerular endothelial cells, but also to podocytes, mesangial cells, and proximal tubules. After binding to cell receptors, the toxin is internalized in the cytosol within 2 hours and inhibits protein synthesis. Treatment of endothelial cells with sublethal doses of Stx, exerting minimal influence on protein synthesis, leads to increased messenger RNA levels and protein expression of chemokines and cell adhesion molecules. By altering endothelial cell adhesion properties and metabolism, Shiga toxins favor leukocyte-dependent inflammation and induce loss of thromboresistance in endothelial cells, leading to microvascular thrombosis.13 Evidence is also emerging that complement activation at the renal endothelial level may contribute to microangiopathic lesions in STEC-HUS. High plasma levels of complement activation products Bb and C5b-9 were measured in children with STEC-HUS, indicating complement activation by the alternative pathway. Stx induced the expression of P-selectin on cultured human microvascular endothelial cell, and P-selectin bound and activated C3 via the alternative pathway, leading to thrombus formation under flow conditions.14 In a murine model of HUS induced by Stx/lipopolysaccharide (LPS), factor B–deficient mice, which cannot activate the alternative pathway of complement, exhibited less thrombocytopenia and were protected against glomerular abnormalities and renal function impairment.14 In vitro, tubular epithelial and mesangial cells are as susceptible to the cytotoxic effects of Shiga toxins as endothelial cells. The tubular damage caused by Stx can lead to a reduction in the renal water-handling capacity. The toxins inhibit water absorption across human renal tubular epithelial cell monolayers, which may contribute to the early events in the pathogenesis of renal dysfunction in STEC-HUS. Diagnosis depends on the detection of E. coli O157:H7 and other STEC and their products in stool cultures. When infection with STEC is suspected, physicians should ensure that stool specimens are collected promptly and specifically cultured for STEC.12 Unlike most other E. coli, serotype O157:H7 does not ferment sorbitol rapidly and thus forms colorless colonies on sorbitol-containing MacConkey agar (SMAC). The use of SMAC provides a simple, inexpensive, and generally reliable method of screening stools for E. coli O157. Suspect colonies can be assayed for the O157 antigen with commercially available antiserum or latex agglutination kits. The use of tests that identify Shiga toxins or the genes encoding them (by polymerase chain reaction) is helpful for diagnosis. Convalescent-phase serum samples can be assayed for antibodies to O157 or other specific strain-derived LPS. However, results may be biased by false-positives caused by antibodies preformed during antecedent STEC exposure.12 E. coli O157:H7 and other STEC have been responsible for multiple outbreaks throughout the world, becoming a public health problem in both developed and developing countries.12 Contaminated undercooked ground beef, meat patties, raw vegetables, fruit, milk, and recreational or drinking water have all been implicated in the transmission of STEC; healthy cattle are a major reservoir for human infection. The large 2011 outbreak of HUS in Germany was caused by ingestion of sprouts contaminated by an unusual STEC strain O104:H4. The chain of transmission appeared to have started in Egypt with fecal contamination of fenugreek seeds by either humans or farm animals. The higher prevalence of women in this outbreak may reflect a gender-specific dietary preference.3 After exposure to STEC, 38% to 61% of individuals develop hemorrhagic colitis, and 3% to 9% (in sporadic infections) to 20% (in epidemic forms) progress to overt HUS12 (Fig. 29-8). STEC-induced hemorrhagic colitis not complicated by HUS is self-limiting and is not associated with an increased long-term risk of high blood pressure or renal dysfunction. STEC-HUS is characterized by prodromal diarrhea followed by acute kidney injury (AKI). The average interval between E. coli exposure and illness is 3 days. Illness typically begins with abdominal cramps and nonbloody diarrhea; diarrhea may become hemorrhagic in 70% of patients, usually within 1 or 2 days.12 Vomiting occurs in 30% to 60% and fever in 30%. Leukocyte count is usually elevated. HUS is usually diagnosed 6 to 10 days after onset of diarrhea. In patients who develop HUS, 70% require RBC transfusions, and 40% to 50% need dialysis for an average 10 days, whereas the remainder have milder renal involvement without the need for dialysis.12,15 About 25% of STEC-HUS patients have neurologic involvement, including lethargy, apnea, cortical blindness, hemiparesis, stroke, seizures, and coma. Rare complications include pancreatitis, diabetes mellitus, and pleural or pericardial effusions. From 1% to 2% of patients die during the acute phase of STEC-HUS. More than 90% of children with STEC-HUS fully recover from the acute disease. However, a meta-analysis of 49 published studies (3476 patients, including children and adults, mean follow-up of 4.4 years) describing the long-term prognosis of patients who survived an episode of STEC-HUS reported death or permanent ESRD in 12% and glomerular filtration rate (GFR) less than 80 ml/min/1.73 m2 in 25% of patients.15 Disease presentation and outcome were particularly severe during the STEC O104:H4 German outbreak, in which 53 of the 855 patients with HUS died.3 Compared with previous STEC epidemics, there was a higher incidence of dialysis-dependent AKI patients (20% vs. 6%) and mortality (6% vs. 1%).3 Almost half the patients presented with neurologic symptoms, and 20% had seizures. The severe clinical phenotype was explained by the lack of previous immunity to this novel STEC strain and its exceptional virulence. E. coli O104:H4 not only produces the same Stx as STEC enterohemorrhagic strains, but also has 93% of the genomic sequence of enteroaggregative E. coli strains that form fimbriae, which facilitate adhesion to the intestinal wall. The evolution of E. coli O104:H4 is likely the result of the acquisition by an enteroaggregative strain of E. coli of a Stx-encoding phage from a Stx-producing enterohemorrhagic strain of E. coli. The combination of these two virulence factors would lead to increased gut colonization and thus the release of increased quantities of toxin into the circulation. Moreover, whereas enterohemorrhagic E. coli bacteria are found in the GI tract of ruminants, enteroaggregative E. coli appear to have their reservoir in humans. This might explain why E. coli O104:H4 strain has acquired new resistances to the most common antibiotics used in human disease. Typical pediatric STEC-HUS treatment relies on supportive management of anemia, renal failure, hypertension, and electrolyte and water imbalance. Initiating intravenous isotonic volume expansion as soon as an E. coli O157:H7 infection is suspected, that is, within the first 4 days of illness, even before culture results are available, may limit the severity of AKI and the need for renal replacement therapy.16 Up to 80% of patients receive packed RBCs for symptomatic anemia. Patients with severe STEC-HUS require careful monitoring, including urine output, weight, volume status, cardiovascular/respiratory function, and early signs of CNS or other organ involvement. Bowel rest is important for the enterohemorrhagic colitis associated with STEC-HUS. Antimotility agents should be avoided because these may prolong the persistence of E. coli in the intestinal lumen, increasing patient exposure to its toxin. The use of antibiotics should be restricted to the very limited number of patients presenting with bacteremia. In children with gastroenteritis, antibiotics may increase the risk of HUS 17-fold;17 possibly because antibiotic-induced injury to the bacterial membrane might favor the acute release of large amounts of preformed toxin. Alternatively, antibiotic therapy might give E. coli O157:H7 a selective advantage if these organisms are not as readily eliminated from the bowel as are the normal intestinal flora. Moreover, several antimicrobial drugs, particularly the quinolones, trimethoprim, and furazolidone, are potent inducers of Stx gene expression and may increase the level of toxin in the intestine. An interesting exception may be azithromycin; its use appeared to have some benefit on the duration of bacterial shedding in adult patients from the German O104:H4 epidemic.18 In contrast to E. coli–associated HUS, hemorrhagic colitis and HUS caused by Shigella dysentery type 1 should be treated with antibiotics. Treatment shortens the duration of diarrhea, decreases the incidence of complications, and reduces the risk of transmission by shortening the duration of bacterial shedding. Careful blood pressure control and renin-angiotensin system (RAS) blockade may be particularly beneficial in the long term for those patients who have CKD after an episode of STEC-HUS. Thus, 8 to 15 years of treatment with angiotensin-converting enzyme (ACE) inhibitors after severe STEC-HUS normalized blood pressure, reduced proteinuria, and improved GFR.19 Among newer treatments for Stx-HUS, Stx-neutralizing monoclonal antibodies are the most advanced, including dual antibodies against Stx 1 and 2 (SHIGATEC) given at the time of GI infection; results of a completed Phase 2 clinical trial are forthcoming. Peptides impairing the ability of enterohemorrhagic E. coli to survive under the acidic conditions of the gastric system could halt the disease process at even earlier stages by preventing bacterial intrusion into the gut. Heparin and antithrombotic agents may increase the risk of bleeding and should be avoided. The efficacy of specific treatments in adult patients is difficult to evaluate because most information is derived from uncontrolled series that also may include atypical HUS cases. In particular, no prospective, randomized controlled trials (RCTs) are available to establish definitely whether plasma infusion or exchange offers specific benefit compared with supportive treatment alone (Table 29-2). However, comparative analyses of two large series of patients treated20 or not treated21 with plasma suggest that plasma therapy may dramatically decrease overall mortality of STEC O157:H7–associated HUS. Plasma infusion or exchange should therefore be considered in adult patients with severe AKI and CNS involvement. Table 29-2 Dosing and effectiveness of specific therapies for patients with hemolytic uremic syndrome or thrombotic thrombocytopenic purpura. aHUS, Atypical hemolytic uremic syndrome; STEC, Shiga toxin–producing E. coli.
Thrombotic Microangiopathies, Including Hemolytic Uremic Syndrome
Definitions
Classification of Hemolytic Uremic Syndrome and Thrombotic Thrombocytopenic Purpura
Clinical Presentation
Etiology
Hemolytic Uremic Syndrome
Stx associated
Infections by Shiga toxin (Stx)–producing bacteria
Neuraminidase associated
Infections by Streptococcus pneumoniae
Atypical Hemolytic Uremic Syndrome
Familial
Mutations: CFH, 40%-45%; CFI, 5%-10%; C3, 8%-10%; MCP, 7%-15%; THBD, 9%; CFB, 1%-2%
Sporadic
Idiopathic
Mutations: CFH, 15%-20%; CFI, 3%-6%; C3, 4%-6%; MCP, 6%-10%; THBD, 2%; CFB, <1%
Anti-CFH antibodies: 6%-10%
Pregnancy associated
Mutations: CFH, 40%-50%; CFI, 10%-20%; MCP, 10%; C3, 14%
HELLP syndrome
Mutations: CFH, 10%; CFI, 20%; MCP, 10%
Transplantation (de novo atypical HUS)
Mutations: CFH, 15%; CFI, 16%
Thrombotic Thrombocytopenic Purpura
Congenital
Homozygous or compound heterozygous mutations in ADAMTS13 gene
Idiopathic
Anti-ADAMTS13 autoantibodies
Secondary
Ticlopidine, clopidogrel
Anti-ADAMTS13 autoantibodies (ticlopidine, 80%-90%, clopidogrel, 30%)
HSC transplantation
Unknown; rarely, low ADAMTS13 levels
Malignancies
Unknown; rarely, low ADAMTS13 levels
HIV
HIV; rarely, low ADAMTS13 levels
SLE, APS, and other autoimmune disease
Depends on the specific primary disease
Laboratory Signs
Pathology
Mechanisms, Clinical Features, and Management of Specific Forms of Thrombotic Microangiopathy
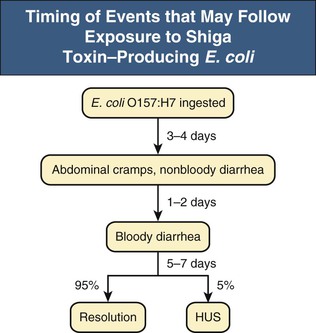
Shiga Toxin–Producing Escherichia coli –Associated Hemolytic Uremic Syndrome
Mechanisms
Diagnosis
Clinical Course
Therapy
Specific Therapies Used in HUS and TTP: Dosing and Efficacy
Therapy
Dosing
Efficacy
Immunosuppressive Agents
Prednisone
200 mg tapered to 60 mg/day, then 5 mg reduction per week
Probably effective in addition to plasma exchange in patients with TTP and anti-ADAMST13 autoantibodies or in aHUS with anti–factor H autoantibodies and in forms associated with autoimmune diseases
Lack of evidence from controlled trials in immune-mediated HUS or TTP
Prednisolone
200 mg tapered to 60 mg/day, then 5 mg reduction per week
Immunoglobulins
400 mg/kg/day
CD20 Cell-Depleting Agent
Rituximab
375 mg/m2/week, up to CD20 depletion
Effective in treatment or prevention of TTP associated with immune-mediated ADAMTS13 deficiency resistant to or relapsing after immunosuppressive therapy
Fresh-Frozen Plasma
Exchange
1-2 plasma volumes/day
First-line therapy for aHUS and TTP
Unproven efficacy in childhood STEC-HUSRelated posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

Thrombotic Microangiopathies, Including Hemolytic Uremic Syndrome
Chapter 29