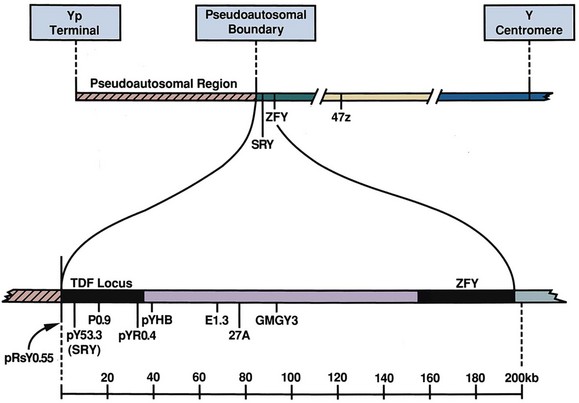
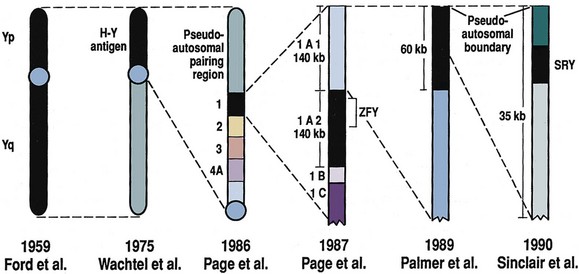
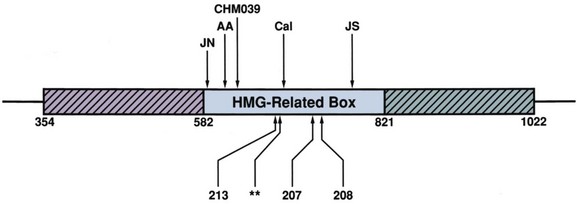
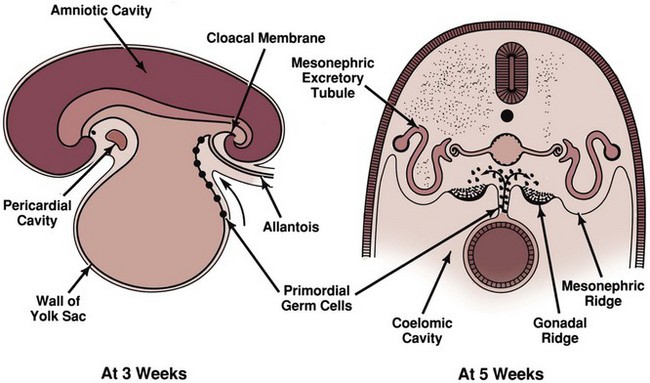
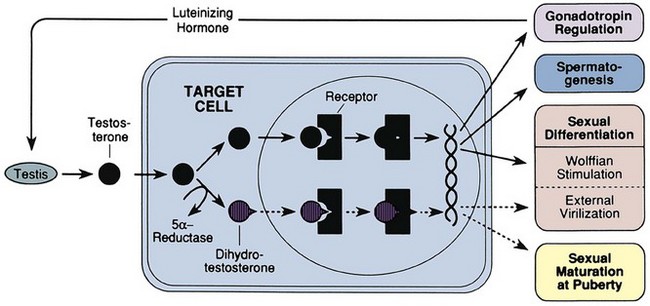
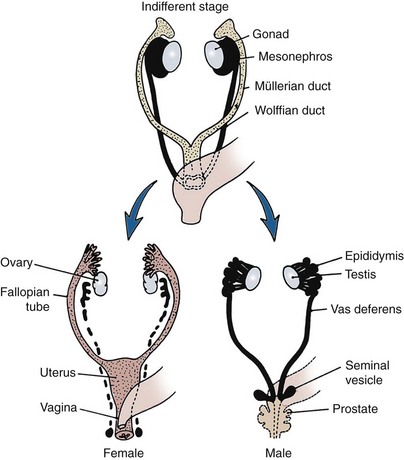
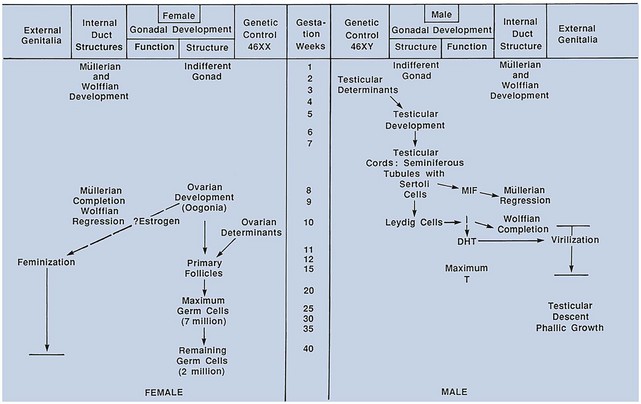
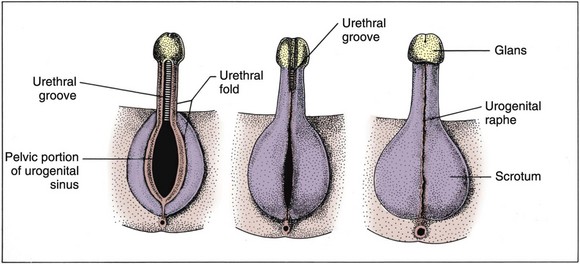
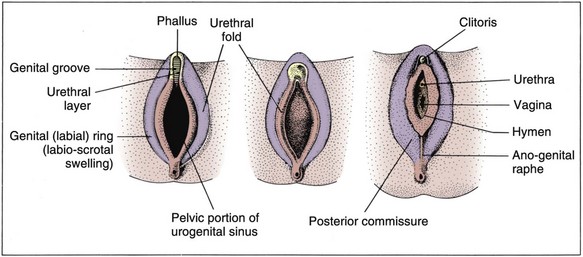
David Andrew Diamond, MD, Richard N. Yu, MD, PhD Under normal circumstances, sexual differentiation is a dynamic and sequential process. According to the Jost paradigm, three steps must occur: establishment of chromosomal sex at fertilization, which determines development of the undifferentiated gonads into testes or ovaries, and subsequent differentiation of the internal ducts and external genitalia as a result of endocrine functions associated with the type of gonad present (Jost et al, 1973). Therefore sexual development occurs as a result of different but complementary processes: genotypic effects, phenotypic events, and gender identity formation. Interference with this highly ordered process at any step can result in a disorder of sexual differentiation. In 1921, Painter demonstrated cytologically that humans have X and Y chromosomes. Based on chromosomal studies of Drosophila, it was assumed that sex was determined by the X chromosomes possessed by the individual (Bridges, 1921). The Y chromosome was thought to impart no genetic information until karyotyping of mammalian chromosomes, developed in the 1950s, demonstrated that the Y chromosome specified development of the testis. Specifically, reports in the late 1950s describing the karyotype 47,XXY as male with Klinefelter syndrome and 45,XO as female with Turner syndrome demonstrated that the presence of a Y chromosome, independent of the number of X chromosomes, resulted in the development of a male embryo, whereas in the absence of a Y chromosome the embryo developed as a female (Ford et al, 1959; Jacobs and Strong, 1959). Therefore, the Y chromosome appeared to possess a gene or genes that determined the destiny of the bipotential gonad as a testis or ovary. In the human, the hypothetical Y-chromosomal gene was termed the testis-determining factor (TDF). Problems with the H-Y antigen theory developed, however. A number of women with 45,X gonadal dysgenesis were found to be H-Y antigen positive. In addition, a mouse model for the male sex reversal syndrome (XX male) was studied in which mice have two X chromosomes and testes owing to a fragment of Y chromosome translocated onto one of the X chromosomes (McLaren et al, 1984). These mice were H-Y antigen negative and azoospermic. As a result of these findings the hypothesis that the H-Y antigen was the product of TDF was excluded. Further study of the Y chromosome suggested that the genetic information responsible for maleness was on the short arm of the chromosome near the centromere. This theory was supported by data from animals with naturally occurring sex chromosome abnormalities. Gain or loss of DNA from the short arm of the Y chromosome on an XX genetic background resulted in either a male or female phenotype, respectively. Further progress was made by studying paradoxical, 46,XX males who develop as phenotypic males in the presence of a presumably normal female 46,XX karyotype (Magenis et al, 1982). The simplest explanation for their sex reversal would be the presence of Y-chromosomal material (including TDF), owing to mosaicism or to submicroscopic cellular quantities. The application of molecular techniques to evaluate Y-chromosomal sequences present in XX males, as well as deletions of the Y chromosome in XY females, led to the cloning of TDF (Lukusa et al, 1992). Deletion maps based on the genomes of these individuals were constructed by a number of laboratories, and TDF was mapped to the most distal aspect of the Y-unique region of the short arm of the Y chromosome, adjacent to the pseudoautosomal boundary (Fig. 133–1). (From Migeon CJ, Berkovitz GD, Brown TR. Sexual differentiation and ambiguity. In: Kappy MS, Blizzard RN, Migeon CJ, editors. Wilkins: the diagnosis and treatment of endocrine disorders in childhood and adolescence. Springfield [IL]: Charles C. Thomas; 1994. p. 588.) However, in subsequent years, data were accumulated that excluded ZFY as the TDF. This included the discovery that in marsupials ZFY was located not on the sex chromosomes but on autosomes (Sinclair et al, 1988). ZFY was excluded with certainty as a candidate for TDF when four individuals with testicular development were found to have inherited a fragment of the Y chromosome that did not include ZFY (Palmer et al, 1989). The renewed search for TDF led to the discovery of Y-specific sequences in XX males lacking ZFY and imposing new limits on the location of TDF to a 35-kb region adjacent to the pseudoautosomal boundary (Fig. 133–2). Using probes from this region, Sinclair and colleagues (1990) discovered a single-copy male-specific sequence that was evolutionarily conserved. This gene was termed SRY (sex-determining region Y gene) in humans and Sry in mice. Analysis of the SRY protein sequence demonstrated a highly conserved, 78 amino acid region with homology to a DNA-binding motif of the high-mobility group (HMG) family of proteins (Fig. 133–3). When DNA encoding the HMG box was used to probe a genomic library, a subfamily of closely related genes was identified. Members of this family, defined as those encoding a region with 60% or greater amino acid similarity to the SRY HMG-box motif, are called SOX (SRY-box-related) genes (Goodfellow and Lovell-Badge, 1993). (From Grumbach MM, Conte FH. Disorders of sex differentiation. In: Wilson JD, Foster DW, editors. Williams textbook of endocrinology. Philadelphia: WB Saunders; 1998. p. 1315.) (From Migeon CJ, Berkovitz GD, Brown TR. Sexual differentiation and ambiguity. In: Kappy MS, Blizzard RN, Migeon CJ, editors. Wilkins: the diagnosis and treatment of endocrine disorders in childhood and adolescence. Springfield [IL]: Charles C. Thomas; 1994. p. 620.) Considerable evidence has accumulated that SRY is the TDF. In the mouse, expression of Sry correlates with testicular determination in the gonadal ridge (Koopman et al, 1990). SRY is an evolutionarily conserved gene on the Y chromosome of mammals. Chromosomal fragments related to SRY (i.e., the SOX genes) are very much conserved evolutionarily, being demonstrated in various vertebrates and marsupials. SRY is localized to the smallest region of the Y chromosome capable of inducing testicular differentiation in humans and in mice (Gubbay et al, 1992). In fact, Koopman and coworkers (1991) introduced into XX mouse embryos a 14-kb mouse genomic DNA fragment containing Sry and no other Y-linked gene sequences and demonstrated that it was capable of giving rise to normal testicular development in the transgenic mice. The SRY protein functions as a transcription factor that, by binding and producing bending of the DNA, promotes protein-protein interaction and is able to activate downstream gene expression. An expectation for TDF was that mutations in its protein sequence would result in sex reversal. Examination of the SRY sequence in XY females has identified more than 20 mutations in the protein-coding sequence. All but one of these point mutations and microdeletions lie within the DNA-binding domain. Another prediction for TDF is that its presence can cause XX male sex reversal. All but one XX male with Y-specific sequences have been found to contain SRY (Goodfellow and Lovell-Badge, 1993). This suggested a causative role of SRY in these cases of XX sex reversal. Therefore, genetic and molecular data have established that SRY can be equated to the TDF. Key Points Chromosomal Sex Several additional genes involved in gonadal development have been identified and characterized. These include, but are not limited to, WT1, SF1, SOX9, DAX1, WNT4, RSPO1, and FOXL2 (Fig. 133–4; Table 133–1). Newer data suggest that these factors function in defined but interrelated differentiation pathways during gonadal formation and determination. Figure 133–4 Genes determining testis or ovarian differentiation from bipotential gonad. (After Hughes IA. Intersex. BJU Int 2002;90:771; and Sekido R, Lovell-Badge R. Sex determination and SRY: down to a wink and a nudge? Trends Genet 2008;25[1]:19–29.) The WT1 gene was originally isolated in cloning experiments that identified an oncogene on human chromosome 11 as being involved in the etiology of Wilms tumor (Call et al, 1990). This gene, originally localized by examining chromosomal deletions in children with WAGR syndrome (Wilms tumor, aniridia, genitourinary abnormalities, gonadoblastoma, and mental retardation), was found to be expressed primarily in the kidney and gonads of the developing human embryo (Kreidberg et al, 1993). The first reported mutations in the Denys-Drash syndrome, which includes Wilms tumor, renal failure, and gonadal and genital abnormalities, were found to involve the WT1 protein (Pelletier et al, 1991a, 1991b). Indeed, mutations involving WT1 have been found responsible for both the Frasier and Denys-Drash syndromes, which appear to represent a spectrum of genetically induced abnormalities involving the gonads and kidneys, owing to the earlier involvement of WT1 in the differentiation of both structures. Frasier syndrome is characterized by both gonadal dysgenesis and renal abnormalities that result in streak gonads and a nephrotic syndrome (MacLaughlin and Donahoe, 2004). If it occurs in the XY genotype, sex reversal results. As a result of alternative splicing of the WT1 gene, patients with Frasier syndrome are not susceptible to Wilms tumor whereas those with Denys-Drash syndrome are (Koziell and Grundy, 1999). In addition, with the Denys-Drash syndrome, gonads differentiate more completely than with Frasier syndrome. Research on Wt1 in the mouse suggests that it exerts its effects upstream of Sry and is likely to be necessary for commitment and maintenance of gonadal tissue (Lim and Hawkins, 1998). Experiments in the mouse have demonstrated that nuclear receptor steroidogenic factor 1 (SF1) is expressed in all steroidogenic tissues, including adrenal cortex, testis (Leydig cells), ovarian theca, granulosa cells, and corpus luteum. SF1 appears to be a key regulator of enzymes involved in steroid production, including the sex hormones, and may directly regulate pituitary gonadotropin expression (Parker et al, 2002). In addition, it appears to play a role in early gonadal development at multiple levels of the reproductive endocrine axis (Ingraham et al, 1994; Luo et al, 1994). SF1 also appears to act synergistically with WT1 in the regulation of müllerian-inhibiting substance (MIS) expression (Shen et al, 1994; Imbeaud et al, 1995; Nachtigal et al, 1998) and may regulate DAX1 expression (Yu et al, 1998). The SOX9 gene was originally identified in patients with camptomelic dysplasia, a congenital disease of bone and cartilage formation that is often associated with XY sex reversal (Wagner et al, 1994). The SOX9 gene is structurally quite similar to SRY, with a 71% sequence similarity of the high-mobility group (HMG) domain to that of SRY. Expression of the gene in adults is greatest in the testes. Interestingly, SOX9 gene activity increases immediately after SRY expression and cell lineage analyses show that SOX9 protein is expressed in cells determined to become Sertoli cells (Sekido and Lovell-Badge, 2009). Therefore, the SOX9 gene may be a key downstream effector of SRY action. The activity of SRY on SOX9 is synergistic with the activity of an additional transcription factor, steroidogenic factor 1 (SF1), which is essential for gonadal and adrenal development (Sekido and Lovell-Badge, 2008). A role for SOX9 in male gonadal determination is further supported by data from genotypic males with SOX9 mutations (46,XY) that exhibit ovarian development and male-to-female sex reversal (Foster et al, 1994; Wagner et al, 1994). SOX9 also upregulates AMH gene expression (MacLaughlin and Donahoe, 2004). The first indication that an X-specific gene was involved in human sex determination was provided in 1978 with the identification of a family with an X-linked mode of inheritance of 46,XY gonadal dysgenesis. Subsequent studies of a number of sex-reversed subjects confirmed the presence of additional X chromosomal genetic material and a normal Y chromosome (Ogata et al, 1992). This finding suggested that duplication of an X-specific gene causes XY sex reversal by expressing a double dose of a region normally subject to X inactivation. Screening of XY females with a normal SRY gene detected such a submicroscopic duplication, involving a 160-kb region designated as the dosage-sensitive sex reversal (DSS) critical region (Bardoni et al, 1994). Parallel studies examining 46,XY males with gonadal dysgenesis, hypogonadotropic hypogonadism, and congenital adrenal hypoplasia resulted in the identification of a candidate gene, NR0B1 (DAX1), within the DSS critical region (Muscatelli et al, 1994; Zanaria et al, 1994). Interestingly, DAX1 can act to suppress or promote the testis determination pathway (Yu et al, 1998; Meeks et al, 2003). Although DAX1 was initially believed to be the primary gene involved in DSS, several sex-reversed 46,XY patients have been identified with duplication of the DSS region in the absence of DAX1 duplication, which suggests that the gene dosage effects of this region may be associated with an additional gene or genes (Zanaria et al, 1995). WNT4 on chromosome 1p34 appears to be involved in müllerian duct regression and may also function by antagonizing SRY activity (Kim et al, 2006; Bernard and Harley, 2007). Early inactivation of Wnt4 in mice causes failure of the formation of müllerian duct derivatives in both sexes. In addition, inactivation of Wnt4 in female mice leads to wolffian duct development without testicular tissue formation and female external genitalia. Furthermore, a recent report supports the role of WNT4 in development and maintenance of the female phenotype in women by regulating müllerian duct development and ovarian steroidogenesis (Biason-Lauber et al, 2004). During the first 6 weeks of embryonic development, the gonadal ridge, germ cells, internal ducts, and external genitalia are bipotential in both 46,XY and 46,XX embryos. Under the genetic influences of sex determination, the bipotential gonadal ridges differentiate into either ovaries or testes and germ cells develop into either oocytes or spermatocytes. Primordial germ cells can be recognized in the third week of gestation on the posterior wall of the secondary yolk sac. Migration of the germ cells begins in the fifth week of gestation as a result of ameboid movements through the mesentery to the medial ventral aspect of the urogenital ridge (Jirasek, 1998) (Fig. 133–5). This process is dependent on chemoattractants and cell adhesion molecules (Hughes, 2002). Overall, a population of 1000 to 2000 primordial germ cells reaches the gonadal blastema by the sixth week of gestation. (From Migeon CJ, Berkovitz GD, Brown TR. Sexual differentiation and ambiguity. In: Kappy MS, Blizzard RN, Migeon CJ, editors. Wilkins: the diagnosis and treatment of endocrine disorders in childhood and adolescence. Springfield [IL]: Charles C. Thomas; 1994. p. 584.) Transformation of the germ cells into spermatogonia and oogonia results from differentiation of the epithelial gonadal compartments referred to as testicular and ovarian “cords.” SRY initiates the switch that induces a cascade of genes directing the indifferent gonad toward testicular organogenesis. The precise moment at which this occurs remains unknown. Initially, differentiation of Sertoli cells is noted as testicular cords form at 6 to 7 weeks’ gestation, creating the basement membrane, or blood-testis barrier, of spermatogonia and Sertoli cells on one side and mesenchymal fibroblasts on the other. The differentiation of Sertoli cells is associated with the production of MIS, a glycoprotein encoded by a gene on the short arm of chromosome 19 (Haqq et al. 1994). In males, a second line of primordial cells of steroidogenic mesenchyme remain among the testicular cords and represent future Leydig cells, which differentiate at 8 to 9 weeks. In the absence of SRY, ovarian organogenesis results. Little is known about the genetic control of ovarian development. To date, no genes whose products direct development of the ovary have been identified. It does appear necessary that there be duplicate copies of at least one X chromosomal locus (which presumably explains the dysgenetic ovaries in the 45,XO Turner syndrome patients). A potential candidate may lie within the DSS critical region on Xp-21, which, when duplicated, promotes male-to-female “sex reversal” (Bardoni et al, 1994; Lopez et al, 1998). Unlike the testis, which functions primarily as a fetal endocrine organ, the ovary has primarily exocrine activity. In embryonal ovaries, germ cells undergo intense mitotic proliferation (preceding the onset of meiotic prophase) and in the process exhaust their entire mitotic potential prenatally, reaching a maximum endowment of 20 million cells by 20 weeks’ gestation. The presence of two X chromosomes appears to be responsible for differentiation of the granulosa cells into the protective mantle of the granulosa layer and “rescue” of 30% of germ cells (approximately 2 million) (Byskov and Westergaard, 1998). Key Point Gonadal State of Differentiation The initial endocrine function of the fetal testes is the secretion of MIS by the Sertoli cells at 7 to 8 weeks’ gestation. MIS, one of the two hormones necessary for male sexual differentiation, acts locally to produce müllerian regression. It is a member of the transforming growth factor-β (TGF-β) family, and the human gene has been cloned and mapped to chromosome 19 (Cate et al, 1986). Little is known about the cellular mechanism of action of MIS. Because the hallmark of MIS-mediated müllerian duct regression is the formation of a ring of connective tissue around the epithelial cells, it is likely that the mesenchyme is the primary target of MIS. Testosterone secretion by the fetal testes is detectable shortly after the formation of Leydig cells in the interstitium at approximately 9 weeks’ gestation (Siiteri and Wilson, 1974). There is a rise in serum and testicular testosterone to a peak concentration at 13 weeks and then a decline. The rate-limiting enzyme for fetal testosterone synthesis is 3β-hydroxysteroid dehydrogenase, which is concentrated approximately 50 times more highly in the fetal testes than in the ovary. Androgens are synthesized by the Leydig cells, initially autonomously, but then dependent on placental human chorionic gonadotropin (hCG) secretion. Later in gestation, with declining hCG concentrations, androgen synthesis is controlled by luteinizing hormone (LH) secretion by the fetal pituitary gland. Jost and colleagues (1973) clearly demonstrated that androgen is essential for virilization of wolffian duct structures, the urogenital sinus, and genital tubercle. Testosterone, the major androgen secreted by the testes, enters target tissues by passive diffusion. Organs such as the wolffian duct, adjacent to the fetal testis, also take up testosterone by pinocytosis. The local source of androgen is important for wolffian duct development, which does not occur if testosterone is supplied only via the peripheral circulation. In some cells, such as those in the urogenital sinus, testosterone is converted to dihydrotestosterone (DHT) by intracellular 5α-reductase. Testosterone or DHT then binds to a high-affinity intracellular receptor protein, and this complex enters the nucleus, where it binds to acceptor sites on DNA, resulting in new messenger RNA and protein synthesis (Fig. 133–6). The androgen receptor has been characterized as a high-affinity receptor that mediates the action of testosterone and DHT in all androgen-dependent tissues. In disorders of the androgen receptor, such as androgen insensitivity syndrome, testosterone production is normal but the hormone is unable to reach the nucleus and interact with DNA. Various defects in the androgen receptor result in a spectrum of phenotypic abnormalities in the genetic male. Because gonadal females have androgen receptor within their tissues, exogenous androgen produces virilization. DHT binds to the androgen receptor with greater affinity and stability than does testosterone. Therefore, in tissues equipped with 5α-reductase at the time of sexual differentiation (e.g., prostate, urogenital sinus, external genitalia), DHT is the active androgen (George and Peterson, 1988). The 5α-reductase activity has two optimal pH values in cultured genital skin fibroblasts—one at pH 5.5 and a second one near pH 8—that correspond to two distinct enzymes (Jenkins et al, 1992). The alkaline enzyme, human steroid 5α-reductase type 1, was cloned first; however, the primary enzyme in the prostate is 5α-reductase type 2 (Andersson and Russell, 1990). A deletion in the gene coding for this enzyme has been discovered in intersex patients with 5α- reductase deficiency (Andersson et al, 1991). The gene encoding the androgen receptor has been cloned and mapped to the X chromosome at Xq11-12 (Lubahn et al, 1988). (From Griffin JE, Wilson JD. Syndromes of androgen resistance. Hosp Pract 1987;22:99–114.) Key Point Gonadal Function Estrogen synthesis is detectable in the female embryo just after 8 weeks of gestation. The rate-limiting enzyme is aromatase, which is higher in the fetal ovary than in the fetal testis. Estrogens are not required for normal female differentiation of the reproductive tract, but they can interfere with male differentiation. Estrogen can block the effect of MIS on müllerian ducts, and prenatal estrogen treatment of mothers has been associated with male reproductive tract abnormalities (Gill et al, 1979; Vigier et al, 1989). Before the 8th week of gestation the urogenital tract is identical in the two sexes. Both the wolffian and the müllerian duct systems are present as anlagen of the internal accessory organs of reproduction (Fig. 133–7). In addition, at this stage, the anlagen of the external genitalia of male and female embryos are indistinguishable (Fig. 133–8). In the male fetus, Sertoli cells produce MIS, which acts locally and unilaterally to suppress the müllerian ducts, and Leydig cells produce testosterone, which permits local development of the wolffian ducts. By 10 weeks of gestation, degeneration of the müllerian ducts is almost complete and the wolffian ducts have become more prominent (see Fig. 133–7). Adjacent to the testes, convolutions of the ducts organize to form the epididymis. The wolffian ducts of the epididymis join with the collecting portion of the testicular tubules (rete testes). Distally, the ducts join the urogenital sinus by about 30 days’ gestation, where they develop into the seminal vesicles. In the female fetus, testosterone is not secreted by the ovaries and therefore the wolffian ducts regress. Because the ovary does not produce MIS, the müllerian ducts are maintained and develop into the female internal reproductive tract. The cephalic ends are anlagen of the fallopian tubes, and the caudal ends fuse to form the uterus (see Fig. 133–7). Contact of the müllerian ducts with the urogenital sinus induces formation of the uterovaginal plate, which ultimately forms the lumen of the vagina. The relative contributions of the müllerian ducts and urogenital sinus to the formation of the vagina remain somewhat controversial; however, there is some agreement that the proximal two thirds of the vagina is contributed by the müllerian ducts and the distal one third by the urogenital sinus. Masculinization of the male fetus starts between 7 and 8 weeks of gestation (Fig. 133–9). The first sign of male phenotypic differentiation is degeneration of the müllerian ducts adjacent to the testes as a result of MIS secretion by the Sertoli cells. Whereas the effects of androgen on the wolffian ducts are related to diffusion of testosterone from the adjacent gonad, masculinization of the external genitalia results from the systemic delivery of testosterone with local conversion to dihydrotestosterone. By 10 weeks, an increase in distance between genital tubercle and anal folds can be seen. The genital tubercle thickens and elongates to become the penis, and the urethral folds fuse from posterior to anterior over the urethral groove (Fig. 133–10). Near the bladder, the urethra is surrounded by the prostate. The urogenital swellings migrate posteriorly to the genital tubercle and fuse to form the scrotum. By 12 to 13 weeks’ gestation, the genitalia of the male fetus is completed with closure of the elongated urogenital cleft. Under the influence of androgen secreted by the fetal testes, penile growth and testicular descent occur in the third trimester (see Fig. 133–9). In the female fetus the absence of circulating testosterone maintains the appearance of the external genitalia at the 6-week gestational stage. The genital tubercle develops only slightly to form the clitoris. The lateral genital swellings become the labia majora, and the adjacent urethral folds become the labia minora (Fig. 133–11). Between the labia minora will develop the vaginal introitus and urethral meatus. Figure 133–7 Differentiation of the wolffian and müllerian duct and urogenital sinus in the male and female. (From Wilson JD. Embryology of the genital tract. In: Harrison HH, Gittes RF, Perlmutter AD, et al, editors. Campbell’s urology. 4th ed. Philadelphia: WB Saunders; 1979. p. 1473.) Figure 133–8 Schematic diagram of external genitalia in the undifferentiated period. (From Martinez-Mora J. Development of the genital tract. In: Martinez-Mora J, editor. Intersexual states: disorders of sex differentiation. Barcelona: Ediciones Doymer; 1994. p. 52.) Figure 133–9 Timetable of normal sexual differentiation. DHT, dihydrotestosterone; MIF, müllerian inhibiting factor. (From Diamond D. Intersex disorders: I and II. AUA Update Series, vol. IX, lessons 9 and 10. Houston: American Urological Association Office of Education; 1990.) Figure 133–10 Schematic diagram of differentiation of the male external genitalia. (From Martinez-Mora J. Development of the genital tract. In: Martinez-Mora J, editor. Intersexual states: disorders of sex differentiation. Barcelona: Ediciones Doymer; 1994. p. 53.) Figure 133–11 Schematic diagram of differentiation of the female external genitalia. (From Martinez-Mora J. Development of the genital tract. In: Martinez-Mora J, editor. Intersexual states: disorders of sex differentiation. Barcelona: Ediciones Doymer; 1994. p. 52.) Humans have been recognized as having sexually dimorphic behavior, which has several aspects: (1) gender identity, the identification of self as either male or female; (2) gender role, aspects of behavior in which males and females appear to differ; (3) gender orientation, or choice of sexual partner (heterosexual, homosexual, or bisexual); and (4) cognitive differences (Grumbach and Conte, 1998). Gender identity is a complex and poorly understood phenomenon in humans, and the mechanisms appear multifactorial. Experience in patients with congenital adrenal hyperplasia (CAH) who were exposed prenatally to androgen and in patients reared in a sex opposite to their chromosomal or gonadal sex have provided evidence to indicate that gender identity is not merely a function of chromosomal complement or prenatal endocrine milieu. Postnatal environmental factors and learning appear to have an important effect. However, strong evidence has accumulated for the impact of prenatal hormonal influences on sexually dimorphic behavior or gender role. For example, long-term follow-up with CAH patients has supported a greater interest in “tomboyish behavior” than in unaffected girls, although these patterns are not abnormal in relation to female behavior in Western society (Ehrhardt and Meyer-Bahlburg, 1981). Additional information regarding the influence of androgens on gender identity and gender role has arisen from one study of males with cloacal exstrophy who underwent gender reassignment within the first few months of life. A majority of these patients had behaviors and attitudes that reflected strong male-typical characteristics regardless of whether they were raised as males or females (Reiner and Gearhart, 2004). However, a subsequent British study of a similar 46,XY cloacal exstrophy cohort presented contradictory findings (Baker Towell and Towell, 2003). The previously accepted dogma that children are psychosexually neutral at birth and capable of being environmentally oriented (the blue room/pink room theory) has been seriously challenged by those who support the concept of prenatal psychosexual differentiation (Money and Ehrhardt, 1972; Diamond and Sigmundson, 1997). Support for either theory in humans is based on the assessment of a limited number of affected patients. An improved understanding of the “nature versus nurture” controversy will probably prove important in the optimal management of patients with DSDs. However, our increased awareness of physiologically normal patients with genuine “gender dysphoria” has illustrated the complexity of this process. Key Point Psychosexual Differentiation The classification of DSDs (previously “intersex disorders”) has undergone evolutionary change as understanding of the etiologic mechanisms of normal and abnormal sexual differentiation has improved. As a result, classification systems vary. The authors have borrowed from the system utilized by Grumbach and Conte (1998), which incorporates the historical emphasis on classification by gonadal morphology and introduced more contemporary terminology (Hughes et al, 2006). Descriptive terms for abnormal gonadal histology have been retained, with the exception of true hermaphroditism, which has been replaced by ovotesticular DSD. But, for masculinized 46,XX females with two ovaries, female pseudohermaphroditism has been replaced by 46,XX DSD. And, for undermasculinized 46,XY males the term male pseudohermaphroditism has been replaced by 46,XY DSD. The first category comprises disorders of gonadal differentiation, the second includes ovotesticular DSD, the third includes 46,XX DSD (the masculinized female, that is, ovaries present but external genitalia exhibiting evidence of masculinization), the fourth includes 46,XY DSD (the undermasculinized male, that is, testes present but genital ducts and/or external genitalia incompletely masculinized), and the fifth category comprises unclassified forms. Within each category, remarkable advances in chromosomal and biochemical information have allowed subclassification of disorders based on etiologic mechanisms, contributing to a more rational classification system (Table 133–2). Table 133–2 Abnormal Sexual Differentiation In 1942, Klinefelter, Reifenstein, and Albright described a syndrome characterized by eunuchoidism, gynecomastia, azoospermia, increased gonadotropin levels, and small, firm testes. By 1959, these patients were noted to have a 47,XXY karyotype (Jacobs and Strong, 1959). Klinefelter syndrome represents the most common major abnormality of sexual differentiation. By definition, males with at least one Y chromosome and at least two X chromosomes have Klinefelter syndrome. The classic 47,XXY complement arises as a result of nondisjunction during meiosis; it occurs in 1 of 600 liveborn males (Morris et al, 2008). But the phenotype is also associated with 48,XXYY and 49,XXXYY, and an exaggerated form of the phenotype is associated with 48,XXXY and 49,XXXXY. The mosaic form 46,XY/47,XXY is associated with a milder version of the phenotypic features of classic 47,XXY Klinefelter syndrome. Perhaps as a result of phenotypic variability, Klinefelter syndrome is believed to be underdiagnosed, with a 25% diagnostic rate in one Danish study (Bojesen et al, 2004). In 47,XXY adults, seminiferous tubules degenerate and are replaced with hyaline. As a result, testes are firm and small, less than 3.5 cm in length. Histologically, Leydig cells appear to be present in large numbers, because they are seen in large clumps in certain areas of the testes, sometimes resembling Leydig cell tumors. However, the absolute volume of Leydig cells is not increased and is probably lower than normal. Serum levels of testosterone are low-normal and those of gonadotropins are elevated. Plasma estradiol levels tend to be high, with gynecomastia the result of an increased ratio of estradiol to testosterone. The vast majority of patients are azoospermic, and the presence of sperm suggests 46,XY/47,XXY mosaicism. There does appear to be a depletion of germ cells with the onset of puberty (Wikström et al, 2004). Fertility, with the benefit of testicular sperm extraction and intracytoplasmic sperm injection, has been reported in patients with Klinefelter syndrome (Koga et al, 2007). Some infertility experts advocate coupling intracytoplasmic sperm injection with preimplantation diagnosis given the lower rate of normal embryos from Klinefelter syndrome patients (54%) versus controls (77%) (Staessen et al, 2003). Gynecomastia, which can be quite marked, is a common pubertal development in patients with Klinefelter syndrome. As a result, these patients have eight times the risk for development of breast carcinoma compared with normal males (Harnden et al, 1971). In addition, they are predisposed to developing malignant neoplasms of extragonadal germ cell origin as well as Leydig and Sertoli cell tumors (Völkl et al. 2006). Therefore, routine surveillance scrotal ultrasonography has been advocated for postpubertal patients with Klinefelter syndrome. An intriguing area of research has been in the neuropsychiatric function of Klinefelter syndrome patients. Studies have demonstrated depressed verbal ability and limitations in frontal executive functioning. Recent imaging studies have shown selective volume differences in corresponding areas of Klinefelter syndrome patients’ brains versus normal subjects (Giedd et al, 2009). Other studies have demonstrated altered cerebral perfusion in Klinefelter syndrome, corresponding to impaired verbal skills (Itti et al, 2003). XX maleness, first recognized by de la Chappelle and coworkers in 1964, is characterized by testicular development in subjects who have two X chromosomes and lack a normal Y chromosome. Most of these subjects have normal male external genitalia, but 10% have hypospadias and all are infertile. Among infertile adults, 2% have XX maleness (Van Dyke et al, 1991). Two categories of patients with XX maleness have been identified: the 90% who are SRY positive and those who are SRY negative (Ergun-Longmire et al, 2005). The SRY-positive group rarely have genital abnormalities, but they have phenotypic features of Klinefelter syndrome, including hypogonadism, gynecomastia, azoospermia, and hyalinization of seminiferous tubules with altered hormonal levels at puberty (low testosterone, increased follicle-stimulating hormone [FSH] and LH) (Fechner et al, 1993). Often, the diagnosis is made in a pubertal male who presents for evaluation of gynecomastia. These patients differ from those with Klinefelter syndrome in that they are shorter (mean height, 168 cm) and have normal skeletal proportions. The 10% of XX males with no detectable SRY more commonly have genital ambiguity. Three mechanisms have been proposed to explain XX sex reversal. The most common is translocation of Y chromosomal material, including SRY, to the X chromosome. Clearly, this can be proven in the majority of patients. Alternatively, sex reversal could result either from the mutation of an autosomal or X chromosomal gene, permitting testicular differentiation downstream from SRY, or from undetected mosaicism with a Y-bearing cell line. Clinical studies have demonstrated XX sex reversal to be a genetically and phenotypically heterogeneous condition (Fechner et al, 1993). In 1938, Henry Turner described the combination of sexual infantilism, webbed neck, and cubitus valgus (increased carrying angle at the elbows) as a distinct entity. Subsequently, gonadal dysgenesis was recognized as part of this syndrome (Hall and Gilchrist, 1990). It was not until 1959 that Ford recognized that one missing X chromosome was the etiologic basis for the syndrome. Subsequent chromosomal studies showed that Turner syndrome is characterized by the presence of only one normally functioning X chromosome. The other sex chromosome may be absent or abnormal, or mosaicism may be present. Turner syndrome has an incidence of 1 in 2500 live births. Half of the patients have a 45,X karyotype in all cells; this is believed to be secondary to loss of an X chromosome through nondisjunction in gametogenesis or an error in mitosis. From 12% to 20% of patients with Turner syndrome have an isochrome X (duplication of one arm of the X chromosome with loss of the other arm). Mosaicism—the presence of two or more chromosomally different cell lines—occurs in 30% to 40% of these patients, the majority (10% to 15%) being 45,X/46,XX and 2% to 5% being 45,X/46,XY (Zinn et al, 1993). The presence of Y chromosomal material is of critical importance in patients with Turner syndrome because it predisposes them to potential masculinization and gonadoblastoma. It is postulated that in Turner syndrome follicular cells that normally surround the germ cells and provide a protective mantle for the oocytes are inadequate (Stanhope et al, 1992). As a result, the rate of attrition of oocytes due to apoptosis is so rapid that by birth few or no oocytes remain in the ovaries, which become streaks (Epstein, 1990). Typically, these streaks are white, fibrous structures, 2 to 3 cm long and approximately 0.5 cm wide, located in the broad ligament. Histologically, the streak possesses interlacing waves of dense fibrous stroma that is devoid of oocytes but is otherwise indistinguishable from normal ovarian stroma. Both estrogen and androgen are decreased, and levels of FSH and LH are increased. Secondary sexual development does not occur in the majority of patients. Pubic and axillary hair fails to develop in normal abundance, and the well-differentiated external genitalia, vagina and müllerian derivatives, and breasts remain small (Saenger, 1996). Turner syndrome is a common cause of primary amenorrhea, and the diagnosis is frequently made because pubertal development never occurs. Spontaneous pubertal development may occur in up to 30% of Turner syndrome patients, however. The associated congenital anomalies that are thought typical in Turner syndrome include short stature, broad chest, widespread nipples, webbing of the neck, peripheral edema at birth, short fourth metacarpal, hypoplastic nails, multiple pigmented nevi, coarctation of the aorta, bicuspid aortic valve, and renal anomalies (Fig. 133–12). The majority of the associated congenital anomalies can be explained by the presence of lymphedema at critical points in development, leading to an imbalance in growth forces. This may be secondary to failed opening of embryonic lymphatic channels (Zinn et al, 1993). (From Diamond D. Intersex disorders: I and II. AUA Update Series, vol. IX, lessons 9 and 10. Houston: American Urological Association Office of Education; 1990.) Of paramount importance in the assessment of the patient with Turner syndrome is identification of Y chromosomal material or 45,X/46,XY mosaicism, whose detection has been enhanced by use of the polymerase chain reaction (PCR) (Bianco et al, 2006). In patients with occult Y chromosomal material, the risk of gonadoblastoma, an in-situ germ cell cancer, is 12% and is being further defined (Schoemaker et al, 2008). Gonadoblastoma is associated with dysgerminoma or other germ cell neoplasms in 50% to 60% of cases, sometimes associated with virilization. Because the age of occurrence of gonadoblastoma is variable and has been reported as early as age 26 months (Peters, personal communication, 2005), timely prophylactic excision of the streak gonads in the Y mosaic Turner syndrome patient is advised. This may be well performed laparoscopically. Streak gonads confirmed to be in 45,XO patients need not be removed. In the recent British national cohort study there was also an increased risk of bladder cancer in Turner syndrome patients followed into adulthood (Schoemaker et al, 2008). Between 33% and 60% of patients with Turner syndrome have structural or positional abnormalities of the kidney; this occurs most frequently in the classic 45,XO karyotype (Hall and Gilchrist, 1990). Horseshoe kidney accounts for 10%, duplication or renal agenesis for 20%, and malrotation for 15% of these abnormalities. Multiple renal arteries have been noted in 90% of patients with Turner syndrome as a result of their cardiovascular evaluation (Hall and Gilchrist, 1990). The contemporary treatment of patients with Turner syndrome has undergone considerable advances. In the neonate, it entails a concerted search for occult Y chromosomal material, including fluorescent in-situ hybridization (FISH) or polymerase chain reaction (PCR) and, subsequently, prophylactic gonadectomy if necessary, as well as ultrasound screening for renal and cardiac abnormalities. In the child, human growth hormone has successfully been employed to achieve increased adult height (Pasquino, 2004). At an appropriate age, typically between 12 and 15 years, exogenous hormonal therapy to induce puberty and then to maintain a normal female endocrine status is begun. An improved understanding of the long-term medical management of these patients, including cardiac surveillance and management of glucose intolerance and osteoporosis, has also resulted in considerable progress. Finally, with the remarkable advances in assisted reproductive technology, pregnancy is a realistic possibility for patients with Turner syndrome, although spontaneous fertility is rare (Sybert and McCauley, 2004). A spectrum of potential gonadal function has been noted in large series of patients with Turner syndrome (Kaneko et al, 1990). In one series, nonstreak gonads were reported in one third of such patients and were more commonly noted in girls with loss of only the short arm of the X chromosome. In 2% to 5% of Turner patients, spontaneous menses will occur with a potential to achieve pregnancy independently (Saenger et al, 2001). This appears most likely in women with mosaicism for a normal 46,XX cell line, a 47,XXX cell line, or distal Xp deletion. To date, more than 160 pregnancies have been reported among spontaneously menstruating Turner syndrome patients. For the vast majority with true streak gonads, for whom egg donor implantation is used, 40% to 50% pregnancy rates have been reported by centers specializing in in-vitro fertilization (Saenger, 1993). However, among these rare pregnancies, the rates of miscarriage, stillbirth, and malformed infants is high (Abir et al, 2001). A recent area of interest has been the laparoscopic retrieval of follicles in girls with Turner syndrome for cryopreservation (Hreinsson et al, 2002). The technique is regarded as experimental and not yet recommended for clinical use. One concern relates to confirmation of normalcy of the harvested oocytes. It is of great interest that neuroanatomic imaging studies of 45,XO Turner syndrome patients have demonstrated differences in parietal and temporal lobe anatomy and posterior fossa morphology that appear to correlate with certain established neurophysiologic and cognitive deficits (Brown et al, 2004; Rae et al, 2004). A familial incidence of 46,XX gonadal dysgenesis has been reported as an autosomal recessive trait (Espiner et al, 1970). This suggests the possibility that autosomal genes in addition to genes on the X chromosome may be involved in ovarian maintenance. The term mixed gonadal dysgenesis was coined by Sohval in 1963. In 1975, Zah and associates reported on their series of more than 100 patients with 45,X/46,XY karyotypes, 72 of whom had mixed gonadal dysgenesis with a streak gonad on one side and a testis on the other. The phenotypic spectrum of patients with XO/XY mosaicism extends from phenotypic females with Turner syndrome (25%), to those with ambiguous genitalia, to, rarely, those appearing as normal males (Berkovitz et al, 1991). In the neonatal period, mixed gonadal dysgenesis is the second most common cause of ambiguous genitalia (after CAH) and must be in the differential diagnosis. The majority of these patients present with varying degrees of phallic development, a urogenital sinus with labioscrotal fusion, and an undescended testis. In virtually all of these patients, a uterus, vagina, and fallopian tube are present. Short stature and associated somatic stigmata are variable features. Key Point Mixed Gonadal Dysgenesis The phenotypic asymmetry of the internal ducts epitomizes the mechanism of local testosterone and MIS production on müllerian and wolffian duct regression and development. In the series of Mendez and colleagues (1993) comprising 16 patients, all had a fallopian tube accompanied by a streak gonad, consistent with absent MIS. Therefore, whereas a dysgenetic or streak gonad is associated with ipsilateral müllerian derivatives (uterus, fallopian tube) (Fig. 133–13), a well-differentiated testis with functional Sertoli and Leydig cells will have ipsilateral wolffian but no müllerian ducts (Davidoff and Federman, 1973). In addition, the presence of severe external genital ambiguity in many of these patients suggests that testosterone production in utero was inadequate to promote complete differentiation of the external genitalia. Paradoxically, the dysgenetic testis is capable of responding to gonadotropins and secreting testosterone in normal quantities at puberty. Yet, despite normal postpubertal endocrine function, it is postulated that fetal testicular endocrine function is either delayed or deficient. Histologically, the testes lack germinal elements, so infertility is the rule. The risk of developing a gonadal tumor (gonadoblastoma, dysgerminoma) is increased in mixed gonadal dysgenesis, with an estimated incidence of 15% to 35% (Robboy et al, 1982; Wallace and Levin, 1990). Gonadoblastoma, a tumor of low malignant potential, is the most common. It was so named because it recapitulates gonadal development more completely than any other tumor (Scully, 1970). Although germ cell tumors occur both in the dysgenetic testes and in the streak gonads of individuals with 46,X/46,XY mosaicism, the risk of tumor is higher in the former (Verp and Simpson, 1987). Patients with mixed gonadal dysgenesis are also at increased risk for Wilms tumor. Rajfer (1981) reported that 50% of 10 patients with an intersex disorder and Wilms tumor had mixed gonadal dysgenesis. He postulated that there was a genetic or teratogenetic defect involving the urogenital ridge, the common embryonic anlage of both kidney and gonad. This concept was borne out by improved understanding of the Denys-Drash syndrome, now clearly associated with mutations in the Wilms tumor suppressor gene (WT1). In 1967, Denys and colleagues described a child with XX/XY mosaicism, nephropathy, genital abnormalities, and Wilms tumor. Drash and coworkers (1970) reported two further examples in 1970. The full triad of the syndrome includes nephropathy, characterized by the early onset of proteinuria and hypertension, and progressive renal failure in most of the patients. Renal histology demonstrates diffuse focal mesangial sclerosis. Because incomplete forms of the syndrome may occur, the nephropathy has become regarded as the common denominator of the syndrome (Habib et al, 1985). Wilms tumor may be diagnosed before, after, or simultaneously with presentation with nephropathy. The majority of the tumors are of favorable triphasic histology (Beckwith and Palmer, 1978). However, there is a high incidence of bilateral Wilms tumor in this syndrome. The genital abnormalities include frank ambiguity, hypospadias, and cryptorchidism. A large number of patients with Denys-Drash syndrome have been noted to have mixed gonadal dysgenesis. These have an increased risk of gonadal tumor of 40% (Table 133–3) (Lee et al, 2006). A relatively new and consistent finding with Denys-Drash syndrome is that of caliceal blunting without obstruction (Jadresic et al, 1990). The high mortality rate associated with this syndrome has prompted an aggressive treatment approach with prophylactic bilateral nephrectomy in an attempt to improve the prognosis for these children (Jadresic et al, 1990). Table 133–3 Risk of Germ Cell Malignancy According to Diagnosis CAIS, complete androgen insensitivity syndrome; PAIS, partial androgen insensitivity syndrome. * Gonadal dysgenesis (including not further specified, 46,XY, 46,X/46,XY, mixed, partial, and complete). † GBY region positive, including the TSPY (testis-specific protein Y encoded) gene. From Lee PA, Houk CP, Ahmed F, et al. Consensus statement on management of intersex disorders. Pediatrics 2006;118:e488–e500. Frasier syndrome, a related disorder due to mutations in the alternative splice donor site of exon 9 on WT1, presents in similar fashion to Denys-Drash syndrome but with certain important distinctions (Klamt et al, 1998). The nephropathy caused by focal segmental glomerulosclerosis occurs later in life with a more gradual progression to renal failure (Koziell et al, 1999). There is no known predisposition to Wilms tumor. Gonadoblastomas in 46,XY individuals are far more common in Frasier syndrome than in Denys-Drash syndrome with a gonadal tumor risk of 60% (see Table 133–3). Because 46,XX individuals with Frasier syndrome have normal gonadal development, they would present with renal failure. But, it is presumed that many such 46,XX individuals go undiagnosed. Frasier syndrome should be considered for girls presenting with steroid-resistant nephrotic syndrome, primary amenorrhea, and pubertal delay (Gwin et al, 2008). The expanded use of prenatal diagnosis has changed the understanding of 45,X/46,XY mosaicism. Studies have shown that 90% to 95% of all infants with 45,X/46,XY mosaicism have normal-appearing male genitalia (Hsu, 1989). Approximately 25% have abnormal gonadal histology (Chang et al, 1990). Because only a small proportion of those with dysgenetic gonads actually have ambiguous genitalia, the possibility exists that some males with gonadal dysfunction have 45,X/46,XY mosaicism. Patients with partial gonadal dysgenesis (dysgenetic male pseudohermaphroditism) are at increased risk for gonadal malignancy. Manuel and colleagues (1976) reported that the incidence of gonadoblastoma or dysgerminoma was 46% by age 40 years. These patients are also at risk for Denys-Drash syndrome (Borer et al, 1995). The etiology of 46,XY complete gonadal dysgenesis may well be an abnormality of the SRY gene that eliminates SRY function, or loss of another gene downstream from SRY that is necessary for SRY protein action. In either case, the absence of testicular determination would permit ovarian differentiation. To date, mutations in the SRY gene are the cause of 46,XY complete gonadal dysgenesis in 10% to 15% of cases. Recently, a mutation in the Desert hedgehog (DHH) gene was noted in 3 of 6 patients with 46,XY complete gonadal dysgenesis, suggesting that the genetic origin of this entity is heterogeneous and the DHH is likely an important gene in gonadal differentiation (Canto et al, 2004). Investigation of a group of individuals with 46,XY complete gonadal dysgenesis has helped to narrow the chromosome interval containing the sex reversal gene to 9p24—a very small region (McDonald et al, 1997). The majority of individuals with 46,XY complete gonadal dysgenesis present in their teens with delayed puberty. In addition to amenorrhea, breast development is usually absent. The serum concentration of gonadotropins is abnormally elevated, which leads the clinician to the determination of karyotype and the subsequent diagnosis (Grumbach and Conte, 1998). The high concentration of serum LH in these patients is thought to be responsible for the increased androgen levels that lead to clitoromegaly in some individuals (Fig. 133–14). (Courtesy of S. Bauer, MD.)
Normal Sexual Differentiation
Normal Genotypic Development
Chromosomal Sex
ZFY and SRY
Additional Genes Involved in Gonadal Determination
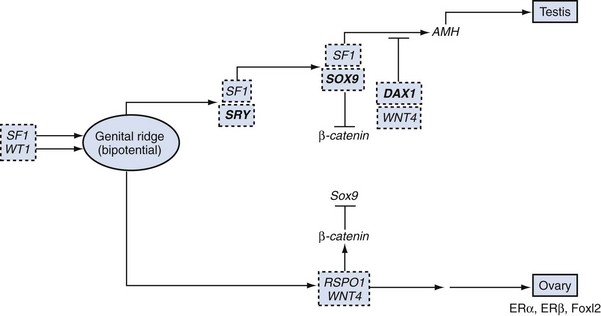
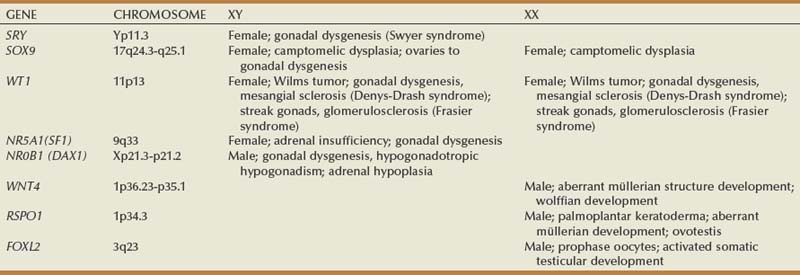
WT1
SF1
SOX9
DAX1 and Dosage-Sensitive Sex Reversal (DSS)
WNT4
Normal Phenotypic Development
Gonadal Stage of Differentiation
Gonadal Function
Ovary
Phenotypic Sexual Differentiation

Gender Identity, Gender Role, and Gender Orientation
Psychosexual Differentiation
Abnormal Sexual Differentiation
1. Disorders of Gonadal Differentiation
2. Ovotesticular DSD (True Hermaphroditism)
3. 46,XX DSD (Masculinized Female)
4. 46,XY DSD (Undermasculinized Male)
5. Unclassified Forms
Disorders of Gonadal Differentiation and Development
Seminiferous Tubule Dysgenesis (Klinefelter Syndrome and Variants)
46,XX Males
Syndromes of Gonadal Dysgenesis
Turner Syndrome
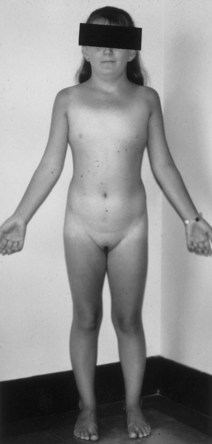
46,XX “Pure” Gonadal Dysgenesis
Mixed Gonadal Dysgenesis
RISK GROUP
DISORDER
MALIGNANCY RISK, %
High
GD*(+Y)† intra-abdominal
15-35
PAIS nonscrotal
50
Frasier
60
Denys-Drash (+Y)
40
Intermediate
Turner (+Y)
12
17β-hydroxysteroid
28
GD (+Y)† scrotal
Unknown
PAIS scrotal gonad
Unknown
Low
CAIS
2
Ovotesticular DSD
3
Turner (-Y)
1
Partial Gonadal Dysgenesis (Dysgenetic Male Pseudohermaphroditism)
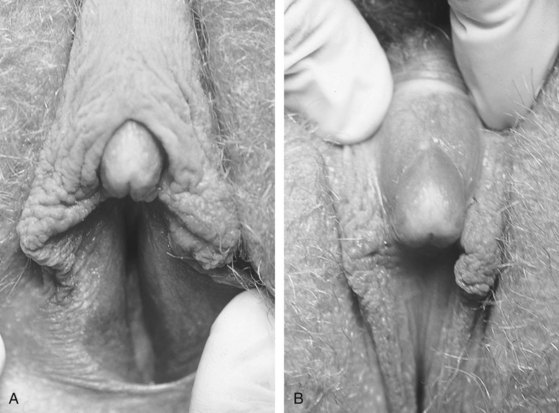
46,XY Complete (Pure) Gonadal Dysgenesis (Swyer Syndrome)
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree