Role of the Kidneys in Hypertension
Joey P. Granger
Eric M. George
Over 40 years ago Guyton and Coleman1,2,3 proposed that if an increase in arterial pressure could produce sustained elevations in sodium and water excretion through a mechanism of renal pressure natriuresis and diuresis, then this system would have a near infinite gain for the long-term control of arterial pressure by regulating blood volume. Thus, whenever arterial pressure is elevated, renal arterial pressure would enhance the excretion of sodium and water until blood volume is reduced sufficiently to return arterial pressure to control values. According to the renal-body fluid system concept, hypertension can develop only when something impairs the excretory ability of the kidney and shifts the relation between sodium excretion and arterial pressure toward higher levels.1,2,3,4 Although there is strong theoretical and experimental evidence that the kidney is a major determinant of the long-term control of arterial pressure, the initial abnormality of the kidney need not be intrinsic to the development of hypertension.4,5,6 A shift of pressure natriuresis can occur as a result of intrarenal abnormalities such as enhanced formation of angiotensin II or genetic defects that enhance renal sodium transport mechanisms. In other instances, the altered kidney function is caused by extrarenal disturbances, such as increased sympathetic nervous system activity or excessive formation of antinatriuretic hormones such as aldosterone.4,5,6
RENAL-BODY FLUID FEEDBACK SYSTEM FOR LONG-TERM BLOOD PRESSURE REGULATION
According to the renal-body fluid feedback mechanism for long-term control of arterial pressure, extracellular fluid volume is determined by the balance between intake and excretion of salt and water by the kidneys (Fig. 39.1). A temporary higher intake than output would lead to an increase in extracellular volume and arterial pressure. If the excretory ability of the kidney is not impaired, the increase in arterial pressure raises sodium excretion and extracellular fluid volume would then decrease, thereby reducing venous return and cardiac output until blood pressure returns to normal and fluid intake and output are reestablished. Conversely, when sodium output exceeds intake and extracellular fluid volume and blood pressure fall below normal, the kidneys retain sodium and water until arterial pressure is restored to the normal set-point. Thus, according to the renal-body fluid feedback mechanism concept, the set-point for long-term blood pressure control is the arterial pressure at which sodium and water intake and output are at equilibrium.4,5,6,7
A key component of this mechanism for regulating salt and water balance is pressure natriuresis/diuresis, which is the effect of increased arterial pressure to raise sodium and water excretion. An important feature of pressure natriuresis is that various hormonal and neural control systems can amplify or blunt the pressure natriuresis mechanism.5,7,9,10,11 For example, in most individuals, chronic increases in sodium intake are associated with only small changes in arterial pressure. The lack of significant increases in arterial pressure in response to elevations in sodium intake results in decreased formation of antinatriuretic hormones and/or increased formation of natriuretic factors, which enhance the effectiveness of pressure natriuresis and allow sodium balance to be maintained with little or no increase in arterial pressure. On the other hand, excessive activation of antinatriuretic systems or abnormalities in natriuretic systems can reduce the effectiveness of pressure natriuresis, thereby necessitating greater increases in arterial pressure to maintain sodium and water balance. Thus, excessive activation of antinatriuretic systems or abnormalities in natriuretic systems impairs the excretory ability of the kidney and shifts the relation between sodium excretion and arterial pressure toward higher levels and resets the set-point for long-term blood pressure control (Fig. 39.2).
Although total peripheral resistance and cardiac output are determinants of arterial pressure, one prediction of the renal-body fluid feedback mechanism is that if the pressure natriuresis mechanism is not impaired, a primary increase in total peripheral resistance or increases in cardiac pumping ability would not result in long-term alterations in arterial pressure.2,3,4,5,6 For instance, an increase in total peripheral resistance would result in an immediate elevation in arterial
pressure. The increase in arterial pressure would increase sodium and water excretion, via pressure natriuresis. As long as fluid excretion exceeds fluid intake, extracellular fluid volume will continue to decrease, reducing venous return and cardiac output, until blood pressure (BP) returns to normal and fluid balance is reestablished. Thus, according to the renal-body fluid volume control system concept, primary increases in total peripheral resistance or increases in cardiac pumping do not result in long-term alterations in arterial pressure and hypertension can develop only when something impairs the excretory ability of the kidney and shifts the relation between sodium excretion and arterial pressure toward higher levels.2,3,4,5,6
pressure. The increase in arterial pressure would increase sodium and water excretion, via pressure natriuresis. As long as fluid excretion exceeds fluid intake, extracellular fluid volume will continue to decrease, reducing venous return and cardiac output, until blood pressure (BP) returns to normal and fluid balance is reestablished. Thus, according to the renal-body fluid volume control system concept, primary increases in total peripheral resistance or increases in cardiac pumping do not result in long-term alterations in arterial pressure and hypertension can develop only when something impairs the excretory ability of the kidney and shifts the relation between sodium excretion and arterial pressure toward higher levels.2,3,4,5,6
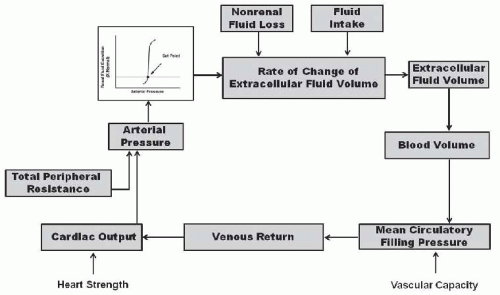 FIGURE 39.1 Basic renal-body fluid feedback mechanism for long-term regulation of blood pressure and body fluid volumes. (Redrawn from Granger JP, Hall JE. Role of the kidney sodium and fluid excretion in hypertension. In: Lip GYP, Hall JE, eds. Comprehensive Hypertension. New York: Elsevier; 2007.) |
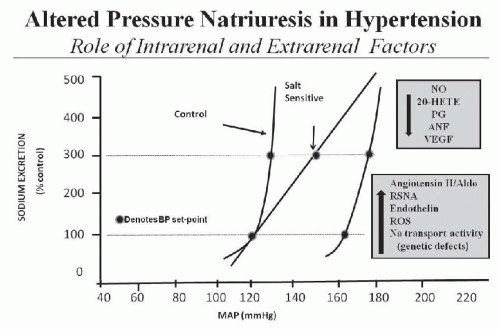 FIGURE 39.2 Steady-state relationships between arterial pressure and urinary sodium excretion and sodium intake for control subjects with normal kidneys and for subjects with a rightward hypertensive shift in the pressure-natriuresis relationship. A shift in the pressure natriuresis relationship can occur as a result of intrarenal abnormalities such as enhanced formation of angiotensin II (ANGII), reactive oxygen species (ROS), inflammatory cytokines and endothelin (via ETA receptor activation), or decreased synthesis of nitric oxide (NO), natriuretic prostanoids (PG), vascular endothelial growth factor (VEGF), or even genetic defects that enhance renal sodium transport mechanisms. In other instances, the altered kidney function is caused by extrarenal disturbances, such as increased sympathetic nervous system (SNS) activity or excessive formation of antinatriuretic hormones such as aldosterone (Aldo) or decreased atrial natriuretic peptide (ANP). |
Although hypertension is a result of a reduction in the kidney’s ability to excrete sodium and water (reduced pressure natriuresis), the hypertension may not necessarily be associated with increases in extracellular fluid volume. Indeed, many forms of hypertension are associated with increased total peripheral resistance and reduced rather than increased extracellular fluid volume. This occurs when a vasoconstrictor, such as norepinephrine, has a potent extrarenal vasoconstrictor effect to increase total peripheral resistance but a relatively weak antinatriuretic action. The antinatriuretic effect of norepinephrine shifts pressure natriuresis and the set-point for arterial pressure to a higher level. However, because norepinephrine has a more powerful extrarenal vasoconstrictor effect, arterial pressure initially increases above the renal set-point for sodium balance and causes transient natriuresis and decrease in extracellular fluid volume. Eventually arterial pressure stabilizes at a point where sodium intake and output are balanced. Thus, sodium retaining actions of norepinephrine are masked by peripheral vasoconstriction which raises BP above the renal set-point at which sodium balance is maintained causing increased renal excretion and decreased extracellular fluid volume. It is important to reiterate that the maintenance of high blood pressure chronically depends on norepinephrine’s renal actions because norepinephrine’s extrarenal effects to increase total peripheral resistance would in itself, as pointed out previously, only result in a transient increase in blood pressure.
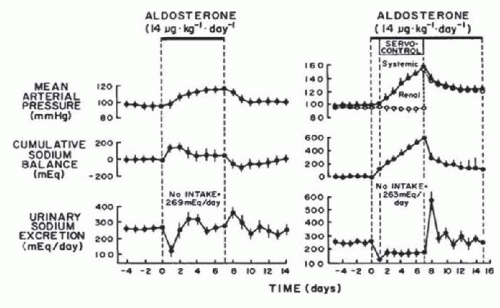 FIGURE 39.3 Effects of chronic aldosterone infusion on sodium excretion when renal perfusion pressure was allowed to increase (left panel) and was servo-controlled (right panel). Notice that when renal perfusion pressure was prevented from increasing, “escape” from sodium retention did not occur and cumulative sodium balance and systemic arterial pressure continued to increase. (Redrawn from Hall JE, Granger JP, Smith MJ, et al. Role of renal hemodynamics and arterial pressure in aldosterone “escape.” Hypertension. 1984, 6(suppl I): I-183-I-192.) |
PRESSURE NATRIURESIS: A KEY FACTOR IN MAINTAINING SODIUM BALANCE IN HYPERTENSION
Another important prediction of the renal-body fluid feedback control system concept is that an increase in BP in hypertensive states is an essential compensatory mechanism that allows sodium balance to be maintained in the face of an underlying sodium retaining defect.5,6,7 The importance of the pressure-natriuresis mechanism in the regulation of sodium excretion can best be illustrated under conditions in which a sodium-retaining abnormality exists within the kidney and an increase in arterial pressure occurs to compensate for this abnormality. Such a condition is illustrated in the hypertension caused by aldosterone excess. To determine the importance of the pressure-natriuresis mechanism in achieving sodium balance during aldosterone-induced hypertension, Hall and colleagues12 examined the long-term effects of aldosterone on sodium excretion and arterial pressure in normal dogs and in dogs in which renal artery pressure was prevented from increasing with an electronically servocontrolled aortic occluder. In dogs in which renal artery pressure was permitted to increase during chronic aldosterone infusion, sodium excretion decreased markedly on the first day and then returned to control levels on days 2 to 3 of aldosterone infusion as arterial pressure increased (Fig. 39.3). In contrast, in dogs in which renal artery pressure was prevented from increasing, sodium excretion decreased on the first day and remained below sodium intake for the 7 days of aldosterone infusion. The sustained sodium retention resulted in dramatic increases in cumulative sodium balance and systemic arterial pressure. The results from this study clearly demonstrated that an increase in renal arterial pressure is essential in allowing the kidneys to escape from the chronic sodium-retaining actions
of aldosterone and to achieve normal sodium balance. Similar findings were reported from the same group during chronic administration of other sodium-retaining hormones, such as angiotensin II and norepinephrine.13,14 Thus, it appears that during pathophysiologic conditions associated with excess levels of sodium-retaining hormones, such as aldosterone or angiotensin II, the pressure-natriuresis mechanism is important for the maintenance of sodium balance.15,16
of aldosterone and to achieve normal sodium balance. Similar findings were reported from the same group during chronic administration of other sodium-retaining hormones, such as angiotensin II and norepinephrine.13,14 Thus, it appears that during pathophysiologic conditions associated with excess levels of sodium-retaining hormones, such as aldosterone or angiotensin II, the pressure-natriuresis mechanism is important for the maintenance of sodium balance.15,16
MECHANISM UNDERLYING RENAL PRESSURE NATRIURESIS
The ability of the kidney to alter urine flow and sodium excretion in response to acute changes in renal perfusion pressure has been of interest to investigators for many years.17,18,19 Changes in sodium excretion in response to changes in renal perfusion pressure are thought to be due to alterations in tubular reabsorption of sodium, because glomerular filtration rate (GFR) and the filtered load of sodium are usually well autoregulated.20 Thus, a decrease in renal perfusion pressure results in an increase in tubular reabsorption of sodium and a decrease in sodium excretion. In contrast, increases in renal perfusion pressure lead to decreases in tubular reabsorption of sodium and increases in sodium excretion, a phenomenon commonly referred to as pressure natriuresis.
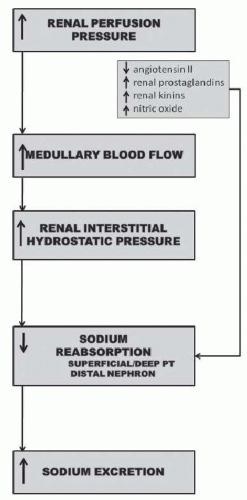 FIGURE 39.4 Mechanisms whereby increases in renal perfusion pressure enhance sodium excretion. |
The specific intrarenal mechanism responsible for the decrease in tubular reabsorption in response to increases in renal perfusion pressure appears to be related to increases in hemodynamic factors such as medullary blood flow and renal interstitial hydrostatic pressure (Fig. 39.4).20,21,22,23,24,25 The mechanism whereby renal interstitial hydrostatic pressure (RIHP) increases in the absence of discernible changes in whole kidney renal blood flow and peritubular capillary hydrostatic and/or oncotic pressures may be related to increases in renal medullary flow as a result of nitric oxide-induced reductions in renal medullary vascular resistance.26 Preventing RIHP from increasing in response to increases in renal perfusion pressure markedly attenuates pressure natriuresis.24 Furthermore, direct increases in RIHP, comparable to increases measured in response to increases in renal perfusion pressure, have been shown to significantly decrease tubular reabsorption of sodium in the proximal tubule and increase sodium excretion.20,21,22,23,24 The exact mechanism whereby RIHP influences tubular reabsorption is unknown but may be related to alterations in tight junction permeability to sodium in proximal tubules, redistribution of apical sodium transporters, and/or release of renal autacoids such as prostaglandin E2.27,28,29,30,31
MECHANISMS OF IMPAIRED RENAL-PRESSURE NATRIURESIS IN HYPERTENSION
Several lines of evidence support an important role for the kidneys in the development and maintenance of hypertension. Some of the strongest evidence for a key role of the kidneys in hypertension derives from renal cross-transplantation studies.4 Transplantation of a kidney from a hypertensive donor into a normotensive recipient has been demonstrated to produce sustained elevations in arterial pressure in numerous genetic models of hypertension (e.g., SHR, Dahl).32 Of particular relevance to human hypertension is the study of Curtis et al.33 demonstrating that BP returns to normal levels in hypertensive patients who were recipients of kidneys from normotensive donors. Another observation that points toward abnormal kidney function as a key factor in causing hypertension is that almost all forms of experimental hypertension are caused by perturbations to the kidneys that alter renal hemodynamics or tubular reabsorption and reduce the kidney’s ability to excrete sodium and water. For example, constriction of the renal arteries (e.g., Goldblatt hypertension), compression of the kidneys (e.g., perinephritic hypertension), or administration of sodium-retaining hormones (e.g., ANG II) are all associated with either initial reductions in renal blood flow and GFR or increases in renal tubular reabsorption prior to development of hypertension.7,34 Further evidence
supporting an important role for the kidneys in the development and maintenance of hypertension is that in all known monogenic forms of human hypertension, the common pathway to hypertension appears to be increased renal tubular reabsorption caused by mutations that directly increase renal electrolyte transport (e.g., Liddle or Gordon syndromes) or the synthesis and/or activity of antinatriuretic hormones (e.g., glucocorticoid remediable aldosteronism).8
supporting an important role for the kidneys in the development and maintenance of hypertension is that in all known monogenic forms of human hypertension, the common pathway to hypertension appears to be increased renal tubular reabsorption caused by mutations that directly increase renal electrolyte transport (e.g., Liddle or Gordon syndromes) or the synthesis and/or activity of antinatriuretic hormones (e.g., glucocorticoid remediable aldosteronism).8
Although specific abnormalities of kidney function are difficult to identify in most patients with primary hypertension, the one aspect of kidney function that is abnormal in all types of experimental and clinical hypertension is renal pressure natriuresis.4,5,7,15,16,34 A shift in the pressure natriuresis relationship can occur as a result of intrarenal abnormalities such as enhanced formation of angiotensin II, reactive oxygen species (ROS), inflammatory cytokines, and endothelin (via ETA receptor activation) or decreased synthesis of nitric oxide and natriuretic prostanoids or even genetic defects that enhance renal sodium transport mechanisms (Fig. 39.2). In other instances, the altered kidney function is caused by extrarenal disturbances, such as increased sympathetic nervous system (SNS) activity or excessive formation of antinatriuretic hormones such as aldosterone. The remaining portion of this chapter discusses how these intra- and extrarenal factors impair renal pressure natriuresis and lead to chronic hypertension.
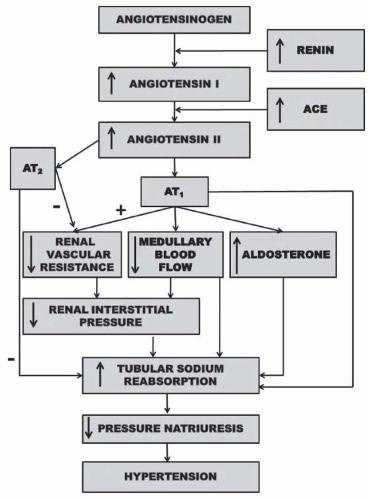 FIGURE 39.5 Renal mechanism whereby activation of the renin-angiotensin system reduces pressure natriuresis relationship and leads to hypertension. |
The Renin-Angiotensin System
The renin-angiotensin system (RAS) plays a critical role in the long-term regulation of blood pressure and is involved in the pathogenesis of various forms of hypertension including renovascular hypertension and human essential hypertension.10,15,34 Although the RAS has many components, its most important effects on renal hemodynamics and sodium reabsorption are exerted by angiotensin II (ANGII) via an ANGII type 1 (AT1) receptor (Fig. 39.5). ANGII also acts on ANGII type 2 receptors to cause renal vasodilation and natriuresis; however, the relative importance of this antihypertensive pathway is equivocal.35,36,37 More recently, fragments of ANGII such as ANG 1-7 have also been suggested to have physiologic effects within the kidney, often opposing the actions of ANGII.35,36,37 An isoform of angiotensin-converting enzyme (ACE), known as ACE2, appears to be involved in the formation of angiotensin 1-7.35,36,37 Although experimental findings on Ang 1-7 are equivocal, ANG 1-7 has been shown to induce renal vasodilation, an effect that is thought to occur independently of binding to AT1 or AT2 receptors
but rather through a G protein-coupled Mas receptor.35,36,37 Although ANG 1-7, ACE2, and the Mas receptor have all been detected within the kidney and an imbalance of these peptides, enzymes, or receptors have been reported to occur in various cardiovascular and renal diseases, the physiologic and pathophysiologic importance of ANG 1-7 has yet to be fully elucidated.35,36,37
but rather through a G protein-coupled Mas receptor.35,36,37 Although ANG 1-7, ACE2, and the Mas receptor have all been detected within the kidney and an imbalance of these peptides, enzymes, or receptors have been reported to occur in various cardiovascular and renal diseases, the physiologic and pathophysiologic importance of ANG 1-7 has yet to be fully elucidated.35,36,37
The RAS, via AT1 receptor, plays an important role in maintaining sodium balance and a relatively normal pressure as sodium intake is altered from low to high levels.9 As sodium intake is increased to high levels, ANGII levels are suppressed allowing sodium excretion to increase to match sodium intake. Conversely, when sodium intake is restricted ANGII levels increase and sodium excretion is reduced to match the low sodium intake. Thus, with a fully functional RAS, sodium balance is maintained in response to changes in sodium intake without the need to invoke significant changes in blood pressure. An inability to suppress the RAS in response to increases in sodium intake could be one potential mechanism for salt-sensitive hypertension in humans.7,10
The importance of the RAS in controlling blood pressure during changes in sodium intake is highlighted by the study of Hall and colleagues9 where they reported that blockade of the RAS, with ANGII receptor blockers (ARB) or ACE inhibitors, increases renal excretory capability so that sodium balance can be maintained at reduced BPs (Fig. 39.6). However, blockade of the RAS also reduces the slope of pressure natriuresis and makes BP salt-sensitive.9 Inappropriately high levels of Ang II reduce renal excretory capability and impair pressure natriuresis, thereby reducing the slope and necessitating increased blood pressure to maintain sodium balance.
The effect of ANGII to reduce renal pressure natriuresis and cause hypertension is the result of its effects to directly or indirectly stimulate sodium transport (Fig. 39.5). Infusion of a physiologic dose of ANGII is usually accompanied by an increase in renal vascular resistance and filtration fraction which favors tubular reabsorption.10,38,39 In addition, acute administration of an ANGII antagonist or converting enzyme inhibitor under conditions where BP is not dramatically reduced results in a natriuresis that is associated with increases in renal blood flow and a reduction in filtration fraction.10 ANGII has been shown to reduce peritubular capillary pressure, whereas converting enzyme inhibition increases peritubular capillary and renal interstitial hydrostatic pressure.10 Thus, alterations in Starling forces across the peritubular capillaries provide an important mechanism whereby the RAS affects the tubular reabsorption of sodium.
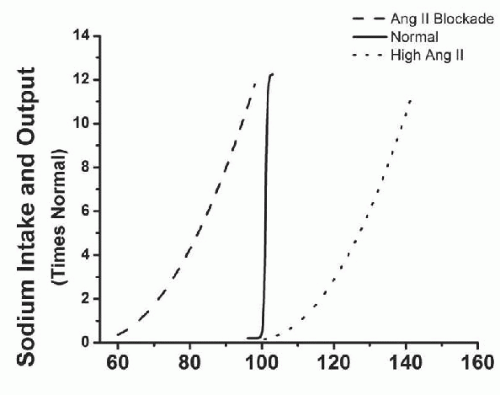 FIGURE 39.6 Effect of changes in mean arterial pressure during chronic changes in sodium intake after angiotensin-converting enzyme (ACE) inhibition, or when angiotensin II was infused at a constant low dose (5 ng/kg/min) to prevent angiotensin II from being suppressed when sodium intake was raised. (Redrawn from data in Hall JE, Guyton AC, Smith MJ Jr, et al. Blood pressure and renal function during chronic changes in sodium intake: Role of angiotensin. Am J Physiol. 1980;239:F271-F280.) |
Alterations in medullary blood flow may be another renal hemodynamic mechanism whereby ANGII could influence tubular reabsorption of sodium.7,10,40 Ang II-induced constriction of efferent arterioles of the juxtamedullary nephrons or pericytes of descending vasa recta would decrease inner medullary blood flow. This, in turn, would enhance sodium reabsorption by increasing medullary interstitial tonicity. In support of this mechanism are studies demonstrating that ANGII, at a dose that does not alter whole-kidney GFR or renal blood flow, markedly reduces medullary blood flow and sodium excretion.10 Studies by Cowley and associates have also shown that inner medullary blood flow is reduced in sodiumdepleted dogs, a condition associated with enhanced activity of the RAS.40 Moreover, ANGII blockade has been shown to improve medullary blood flow and enhance pressure natriuresis in various animal models of hypertension.7,10,40
Experimental evidence also suggests an important direct tubular action of ANGII.10,38,39 Various studies suggest that increased proximal tubule transport in response to physiologic concentrations of ANGII is mediated by changes in HCO3- coupled with Na transport.38 This effect of ANGII has been attributed to activation of Na/H exchange. In addition to a proximal tubule effect on sodium transport, evidence also indicates ANGII may have direct actions on more distal parts of the nephron.10,38,39
Although AT1 receptors are prominently expressed in the kidney, they are also expressed in the heart, blood vessels, adrenal glands, and the brain.10 Because AT1 receptors are ubiquitously expressed, dissecting the quantitative importance of each individual organ system, including the kidney, in the long-term regulation of blood pressure has been difficult. Utilizing a combined gene targeting with renal cross transplantation approach, Coffman and colleagues examined the role of AT1 receptors in the kidney and their contribution to the development of ANGII-induced hypertension.41,42,43,44 They found that ANG II causes hypertension primarily through effects on AT1 receptors in the kidney associated with reduced urinary sodium excretion, independent of actions of the sympathetic nervous system or aldosterone. When AT1 receptors are eliminated from the kidney, the extrarenal AT1 receptors are not sufficient to induce hypertension (Fig. 39.7). Thus, despite the fact that ANGII activates extrarenal vascular receptors and increases total peripheral resistance, when AT1 receptors are eliminated from the kidney, ANGII does not alter the pressure natriuresis relationship or increase arterial pressure.
More recently Coffman and colleagues reported that abrogation of AT1 receptors in the proximal tubule alone reduces proximal fluid reabsorption, alters expression of key sodium transporters, improves pressure-natriuresis, and significantly attenuates ANGII hypertension.42 Collectively, the findings of Coffman and colleagues highlight the critical role of the kidney in the pathogenesis of ANGII-dependent hypertension. In addition, they suggest that the major mechanism of action of RAS inhibitors in hypertension is attenuation of ANG II effects in the kidney.
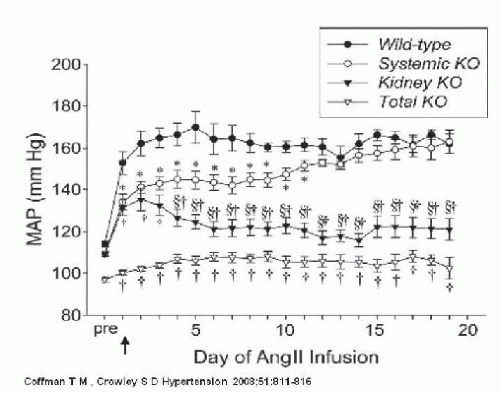 FIGURE 39.7 Blood pressures in cross-transplanted mice during 21 days of AngII infusion. Blood pressure response to AngII in systemic knockout (KO) recapitulates that of the wild-type group by day 12 of AngII infusion. Absence of renal AT1A receptors in kidney KO animals ameliorates AngII-induced hypertension.Total KO blood pressure shows minimal response to AngII infusion. (Coffman TM, Crowley SD. Kidney in hypertension: Guyton redux. Hypertension. 2008;51(4):811-816.) |
Aldosterone
Aldosterone also plays an important role in the chronic regulation of BP via its sodium-retaining actions on the kidney.10,45,46,47 Aldosterone alters the renal pressure natriuresis relationship by enhancing sodium transport in the distal tubules and cortical collecting ducts. The sodium retaining effect of aldosterone is due to binding of aldosterone to the intracellular mineralocorticoid receptor and activation of transcription by target genes. These target genes, in turn, stimulate synthesis or activation of the sodium-potassium ATP-ase pump on the basolateral epithelial membrane and activation of amiloride-sensitive sodium channels on the luminal side of the epithelial membrane.47,48
As sodium intake is increased to high levels, aldosterone levels are suppressed allowing sodium excretion to increase to match sodium intake. Conversely, when sodium intake is restricted aldosterone levels increase and sodium excretion is reduced to match the low sodium intake. Thus, a change in aldosterone production in response to changes in sodium intake is another important hormone in the maintenance of sodium balance. An inability to suppress aldosterone production in response to increases in sodium intake therefore is another potential mechanism for salt-sensitive hypertension in humans.46 In addition to primary hyperaldosteronism, excess activation of the mineralocorticoid receptor by aldosterone has also been implicated in several forms of human hypertension including renovascular hypertension, patients with resistant hypertension, and obesity-related hypertension.46,47,48,49,50,51
The Sympathetic Nervous System
Another antinatriuretic system that can reduce the renal pressure natriuresis relationship and cause chronic hypertension is the renal sympathetic nervous system.52,53,54,55,56 The kidneys receive extensive sympathetic innervation and increases in renal sympathetic nerve activity reduce sodium excretion by increasing tubular reabsorption or decreasing the filtered load of sodium via alpha adrenergic receptor activation (Fig. 39.8).52 Renal nerves can act directly on the tubule to increase sodium reabsorption or indirectly by increasing renal vascular resistance and reducing medullary blood flow and renal interstitial pressure. In addition, increased renal sympathetic nerve activity can enhance tubule reabsorption by activating the RAS.
Excessive activation of the renal SNS has been implicated in the pathogenesis of several experimental and genetic forms of hypertension.52,53




Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
