Horacio J. Adrogué, Nicolaos E. Madias
Respiratory Acidosis, Respiratory Alkalosis, and Mixed Disorders
Deviations of systemic acidity in either direction can have adverse consequences and, when severe, can be life threatening to the patient. Therefore, it is essential for the clinician to recognize and properly diagnose acid-base disorders, to understand their impact on organ function, and to be familiar with their treatment and the potential complications of treatment.1,2
Respiratory Acidosis (Primary Hypercapnia)
Definition
Respiratory acidosis is the acid-base disturbance initiated by an increase in carbon dioxide tension (Pco2) of body fluids and in whole-body CO2 stores. The secondary increment in plasma bicarbonate ion concentration (plasma [HCO3−]) observed in acute and chronic hypercapnia is an integral part of the respiratory acidosis.3 The level of arterial CO2 tension (Paco2) is greater than 45 mm Hg in patients with simple respiratory acidosis, as measured at rest and at sea level. (To convert values from millimeters Hg to kilopascals [kP], multiply by 0.1333.) An element of respiratory acidosis may still occur with lower Paco2 in patients residing at high altitude (e.g., 4000 m or 13,000 ft) or with metabolic acidosis, in whom a normal Paco2 is inappropriately high for this condition.4 Another special case of respiratory acidosis is the presence of arterial eucapnia, or even hypocapnia, occurring together with severe venous hypercapnia, in patients having an acute, profound decrease in cardiac output but relative preservation of respiratory function.5,6 This disorder is known as pseudorespiratory alkalosis and is discussed under respiratory alkalosis.
Etiology and Pathogenesis
The ventilatory system is responsible for eucapnia by adjustment of alveolar minute ventilation ( ) to match the rate of CO2 production. Its main elements are the respiratory pump, which generates a pressure gradient responsible for airflow, and the loads that oppose such action.
) to match the rate of CO2 production. Its main elements are the respiratory pump, which generates a pressure gradient responsible for airflow, and the loads that oppose such action.
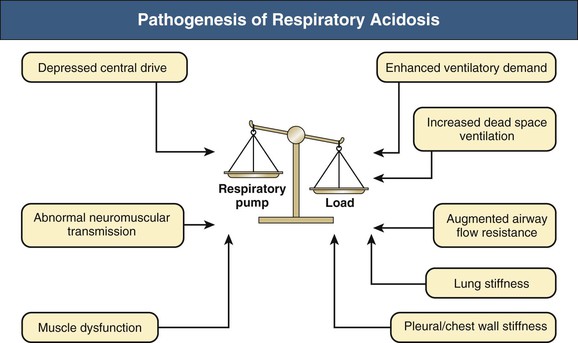
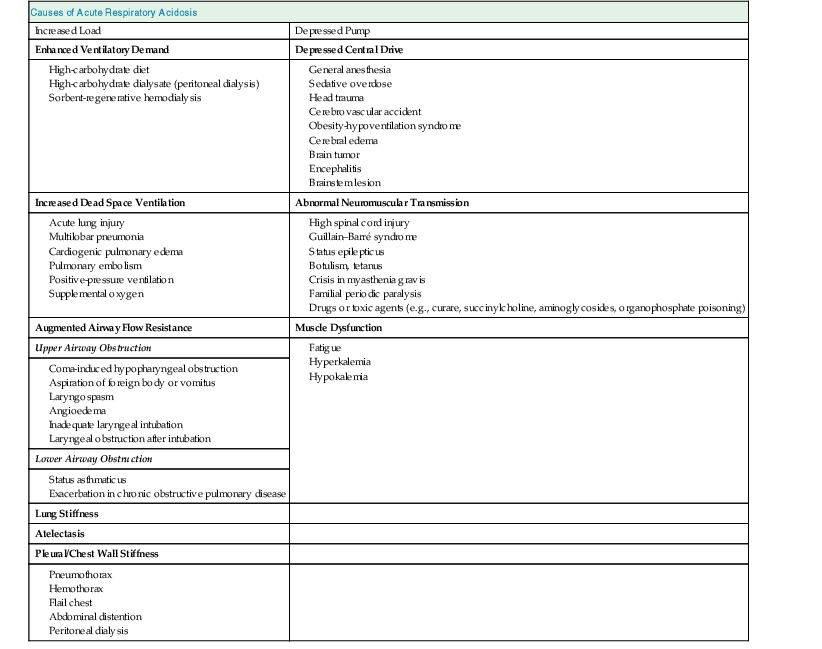
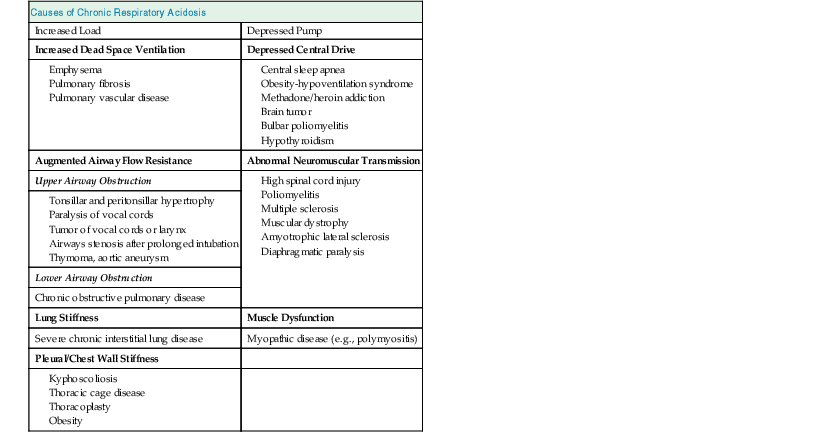
Carbon dioxide retention can occur from an imbalance between the strength of the respiratory pump and the extent of respiratory load (Fig. 14-1). When the respiratory pump is unable to balance the opposing load, respiratory acidosis develops. Respiratory acidosis may be acute or chronic (Tables 14-1 and 14-2). Life-threatening acidemia of respiratory origin can occur during severe, acute respiratory acidosis or during respiratory decompensation in patients with chronic hypercapnia.
A simplified form of the alveolar gas equation at sea level and on breathing of room air (Fio2, 21%) is as follows:
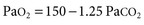
where Pao2 is alveolar O2 tension (mm Hg). This equation demonstrates that patients breathing room air cannot reach Paco2 levels much greater than 80 mm Hg (10.6 kP) because the hypoxemia that would occur at greater values is incompatible with life. Therefore, extreme hypercapnia occurs only during O2 therapy, and severe CO2 retention is often the result of uncontrolled O2 administration.
Secondary Physiologic Response
Adaptation to acute hypercapnia elicits an immediate increment in plasma [HCO3−] due to titration of non-HCO3− body buffers; such buffers generate HCO3− by combining with H+ derived from the dissociation of carbonic acid, as follows:
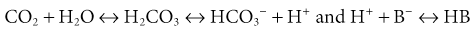
where B− refers to the base component and HB refers to the acid component of non-HCO3− buffers. This adaptation is completed within 5 to 10 minutes from the increase in Paco2, and assuming a stable level of hypercapnia, no further change in acid-base equilibrium is detectable for a few hours.7 Moderate hypoxemia does not alter the adaptive response to acute respiratory acidosis. However, preexisting hypobicarbonatemia (whether it is caused by metabolic acidosis or chronic respiratory alkalosis) enhances the magnitude of the HCO3− response to acute hypercapnia; this response is diminished in hyperbicarbonatemic states, whether caused by metabolic alkalosis or chronic respiratory acidosis.8,9
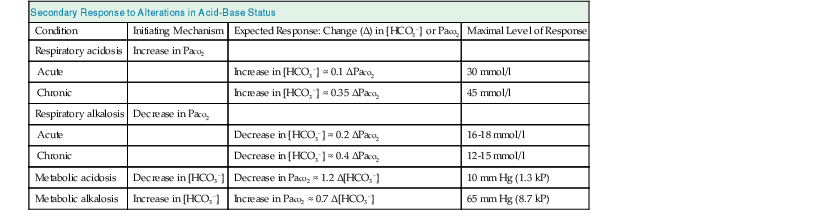
The adaptive increase in plasma [HCO3−] observed in acute hypercapnia is amplified greatly during chronic hypercapnia as a result of HCO3− generation by the kidney.10 In addition, the renal response to chronic hypercapnia includes a reduction in the rate of Cl− reabsorption, resulting in depletion of body Cl− stores. Complete adaptation to chronic hypercapnia requires 3 to 5 days.7 Table 14-3 provides quantitative aspects of the secondary physiologic responses to acute and chronic hypercapnia. More recently, a substantially steeper slope for the change in plasma [HCO3−] in chronic respiratory acidosis was reported (0.51 mmol/l for each 1 mm Hg chronic increase in Paco2), but the small number of arterial blood gas (ABG) measurements, one for each of 18 patients, calls in question the validity of the conclusion reached.11 The renal response to chronic hypercapnia is not altered appreciably by dietary Na+ or Cl− restriction, moderate K+ depletion, alkali loading, or moderate hypoxemia. However, recovery from chronic hypercapnia is crippled by a diet deficient in Cl−; in this circumstance, despite correction of the level of Paco2, plasma [HCO3−] remains elevated as long as the state of Cl− deprivation persists, leading to posthypercapnic metabolic alkalosis.
Clinical Manifestations
Because clinical hypercapnia almost always occurs in association with hypoxemia, it is often difficult to determine whether a specific manifestation is the consequence of elevated Paco2 or reduced Pao2. Nevertheless, the clinician should bear in mind several characteristic manifestations of neurologic or cardiovascular dysfunction to diagnose the condition accurately and to treat it effectively.4,7
Neurologic Symptoms
Acute hypercapnia is often associated with marked anxiety, severe breathlessness, disorientation, confusion, incoherence, and combativeness. A narcotic-like effect can occur in patients with chronic hypercapnia, and drowsiness, decreased alertness, inattention, forgetfulness, loss of memory, irritability, confusion, and somnolence may be observed. Motor disturbances, including tremor, myoclonic jerks, and asterixis, are frequently observed with both acute and chronic hypercapnia. Sustained myoclonus and seizure activity can also develop. Signs and symptoms of increased intracranial pressure (ICP) such as pseudotumor cerebri are occasionally evident in patients with either acute or chronic hypercapnia and appear to be related to the vasodilating effects of CO2 on cerebral blood vessels. Headache is a frequent complaint. Blurring of the optic discs and frank papilledema can be found when hypercapnia is severe. Hypercapnic coma characteristically occurs in patients with acute exacerbations of chronic respiratory insufficiency who are treated injudiciously with high-flow O2.12
Cardiovascular Symptoms
Acute hypercapnia of mild to moderate degree is usually characterized by warm and flushed skin, bounding pulse, sweating, increased cardiac output, and normal or increased blood pressure. By comparison, severe hypercapnia might be attended by decreases in both cardiac output and blood pressure. Cardiac arrhythmias occur frequently in patients with either acute or chronic hypercapnia, especially those receiving digoxin.
Renal Symptoms
Mild to moderate hypercapnia results in renal vasodilation, but acute increments in Paco2 to levels above 70 mm Hg (9.3 kP) can induce renal vasoconstriction and hypoperfusion. Salt and water retention typically attend sustained hypercapnia, especially in the presence of cor pulmonale. In addition to the effects of heart failure on the kidney, multiple other factors might be at play, including the prevailing stimulation of the sympathetic nervous system and the renin-angiotensin-aldosterone axis, the increased renal vascular resistance, and the elevated levels of antidiuretic hormone (ADH) and cortisol.12
Diagnosis
Whenever CO2 retention is suspected, ABG values should be obtained.12 If the patient’s acid-base profile reveals hypercapnia in association with acidemia, at least an element of respiratory acidosis must be present. However, hypercapnia can be associated with a normal or even an alkaline pH if certain additional acid-base disorders are also present. Information from the patient history, physical examination, and ancillary laboratory data should be used to assess whether part or all of the increase in Paco2 reflects an adaptive response to metabolic alkalosis rather than being primary in origin.
Treatment
As previously noted, CO2 retention, whether acute or chronic, is always associated with hypoxemia in patients breathing room air. Consequently, O2 administration represents a critical element in the management of respiratory acidosis.1,13 However, supplemental O2 may lead to worsening hypercapnia, especially in patients with chronic obstructive pulmonary disease (COPD). Although a depressed respiratory drive in CO2 retention seems to play a role, other factors might account for the worsening hypercapnia in response to supplemental O2 therapy. These include an increase in dead space ventilation and ventilation/perfusion (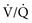 ) mismatch caused by the loss of hypoxic pulmonary vasoconstriction and the Haldane effect (decreased hemoglobin affinity for CO2 in the presence of increased O2 saturation), which mandates an increase in ventilation to eliminate the excess CO2.7
) mismatch caused by the loss of hypoxic pulmonary vasoconstriction and the Haldane effect (decreased hemoglobin affinity for CO2 in the presence of increased O2 saturation), which mandates an increase in ventilation to eliminate the excess CO2.7
Figures 14-2 and 14-3 outline the management of acute respiratory acidosis and chronic respiratory acidosis. Whenever possible, treatment must be directed at removal or amelioration of the underlying cause. Immediate therapeutic efforts should focus on securing a patent airway and restoring adequate oxygenation by delivery of an O2-rich inspired mixture. Supplemental oxygen can be administered to the spontaneously breathing patient with nasal cannulas, Venturi masks, or non-rebreathing masks. Oxygen flow rates up to 5 l/min can be used with nasal cannulas; each increment of 1l/min increases the fraction of inspired O2 concentration (Fio2) by approximately 4%. Venturi masks, calibrated to deliver Fio2 between 24% and 50%, are most useful in patients with COPD because the Po2 can be titrated, thus minimizing the risk of CO2 retention. Oxygen saturation of hemoglobin of approximately 80% to 90% can be achieved with nonbreathing masks.

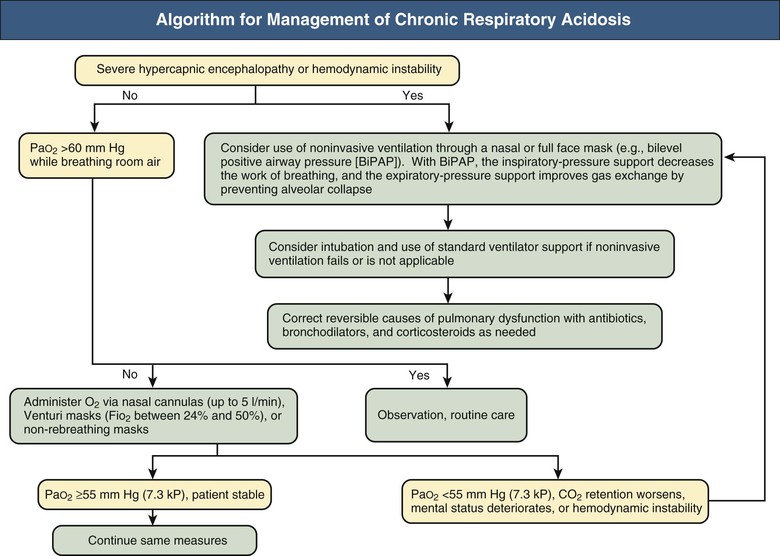
If the target Po2
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


