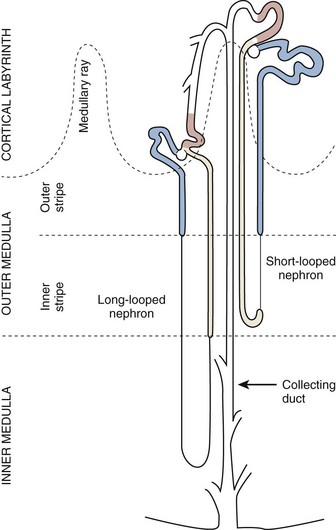
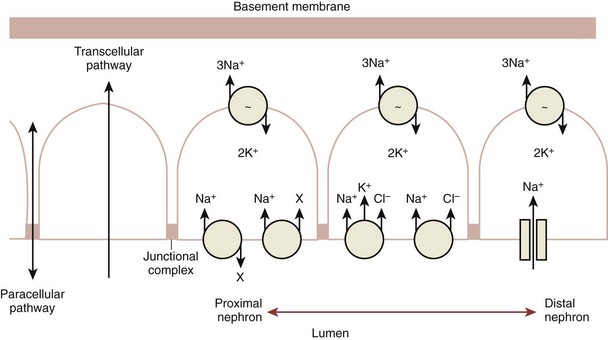
Daniel A. Shoskes, MD, MSc, FRCSC, Alan W. McMahon, MD Under normal resting conditions, RBF is 20% of total cardiac output. Total blood flow is different for men and women, averaging 982 ± 184 mL/min in women and 1209 ± 256 mL/min in men (Dworkin and Brenner, 2004). Renal plasma flow (RPF) is slightly less, averaging 592 mL/min in women and 659 mL/min in men, and varies with hematocrit (RPF = RBF × [1−Hct]). RBF is not evenly distributed to all parts of the kidney. Flow to the outer cortex is 2 to 3 times greater than that to the inner cortex, which in turn is two to four times greater than that to the medulla (Dworkin and Brenner, 2004). where Lp = glomerular permeability and S = glomerular surface area. If all these criteria are met, then: and since and since we can now see that There are a number of mathematical formulas that have been developed to improve the accuracy of the PCr estimation of GFR (National Kidney Foundation, 2002). The two most widely used are the Cockcroft-Gault and “modification of diet in renal disease” (MDRD) formulas. Key Points: Renal Blood Flow and Glomerular Filtration Rate Vascular tone of the renal vessels, the net balance of vasoconstrictive and vasodilatory forces, is crucial to the maintenance of renal blood flow, GFR, tubular renal function, and systemic blood pressure. There is a complex network of hormones and vasoactive substances with both direct and indirect effects, resulting in a system that is pleiotropic and redundant. Although much has been learned from animal models about the function of individual molecules, the complexity of the total system can lead to unexpected outcomes when individual pathways are manipulated pharmacologically. A summary of substances known to impact vascular tone is given in Table 38–1. Table 38–1 Vasoactive Substances That Control Renal Artery Tone Angiotensin II is a potent vasoconstrictor. In the kidney, there is a more pronounced constrictive effect on the efferent than the afferent arteriole, due to inhibition of angiotensin II actions in the afferent arteriole by nitric oxide and prostaglandin (Arima, 2003). Elevated levels of angiotensin II are important for maintaining GFR in pathologic conditions that reduce RBF (e.g., renal artery stenosis, dietary sodium restriction). The classical effects of angiotensin II (vasoconstriction, aldosterone release, sodium retention) are mediated by the AT1 receptor (Kaschina and Unger, 2003). The AT2 receptor, however, may cause intrarenal dilation and be protective against renal ischemic injury (Carey, 2005). Norepinephrine vasoconstricts all the major vascular beds in the kidney, mediated through the α1 receptor. In patients who receive norepinephrine as a pressor agent in the face of systemic vasodilation, renal function is preserved and may actually improve (Albanese et al, 2004). Endothelin is the most potent vasoconstrictor yet identified. There are three isoforms, with ET-1 being the most fully described. An endothelin precursor (big ET-1; 39 amino acids) is cleaved to ET-1 (21 amino acids) by an endothelin converting enzyme found on the endothelial cell membrane. The endothelin receptors are subclassified into ET (A), which are purely vasoconstrictive, and ET (B). The ET (B) receptors may cause either vasodilation by stimulating the release of nitric oxide from endothelial cells, or vasoconstriction of vascular smooth muscle cells (Fellner and Arendshorst, 2004). ET-1 release is stimulated by angiotensin II, antidiuretic hormone, thrombin, cytokines, reactive oxygen species, and shearing forces acting on the vascular endothelium. ET-1 release is inhibited by nitric oxide, as well as by prostacyclin and atrial natriuretic peptide. Blockade of the ET (A) receptor can reduce renal vasoconstriction seen in such ischemic conditions as ureteral obstruction (Bhangdia et al, 2003). ET-1 has a number of other actions besides vasoconstriction. ET-1 stimulates aldosterone secretion, produces positive inotropy and chronotropy in the heart, decreases renal blood flow and GFR, and releases atrial natriuretic peptide. Despite reduction in RBF, sodium excretion is increased, suggesting that ET may be responsible for maintaining sodium balance when the renin-angiotensin system is depressed (Perez del Villar et al, 2005). Medullary blood flow is preserved in the face of endothelin induced vasoconstriction, which may explain the relative stimulation of tubular functions (Evans et al, 2004). Vasopressin acts directly on blood vessels through the vasopressin V1 receptor but does not directly change RBF at low doses (Malay et al, 2004). Vasopressin does potentiate the vasoconstrictive effects of norepinephrine (Segarra et al, 2002) and can induce renal ischemia at high doses. At the low doses typically employed in the management of septic shock, renal function is preserved (Holmes et al, 2001). Atrial natriuretic peptide (ANP) is a vasoactive hormone synthesized primarily by the atria in response to stretching, as occurs during physiologic levels of volume expansion (Fig. 38–2). The primary actions of ANP on the kidney are increased GFR and natriuresis. ANP can increase GFR without a change in renal blood flow (Sward et al, 2005) by the combination of afferent arteriolar vasodilatation and efferent arteriolar vasoconstriction. In addition, ANP dilates vessels that have been preconstricted by norepinephrine, angiotensin II, or vasopressin. ANP production increases during bilateral obstructive uropathy, which may be one mechanism of preserving GFR (Kim et al, 2002). ANP increases natriuresis mainly through inhibition of sodium reabsorption in the medullary collecting duct (Zeidel et al, 1988); decreased renin and decreased aldosterone production may also play a role (Laragh, 1985). Clinically, however, infusion of low-dose ANP during surgery increases water and electrolyte excretion without measured systemic changes in cortisol, angiotensin II, or aldosterone (Koda et al, 2005). This approach has also been used to prevent ischemic renal damage in high-risk cardiac surgery (Sward et al, 2004). Nitric oxide (NO) is a highly reactive gas that participates in multiple physiologic and pathophysiologic reactions in the body. NO is synthesized from the reaction between arginine, reduced nicotinamide adenine dinucleotide phosphate (NADPH), and oxygen to produce citrulline, NADP, water, and NO. This reaction is catalyzed by a family of enzymes called nitric oxide synthase (NOS). Although all NOS enzymes catalyze the same reaction, they differ in distribution, expression, and stimuli. Neuronal (nNOS, NOS-1) and endothelial (eNOS, NOS-3) are constitutively expressed, and iNOS (NOS-2) is inducible. eNOS is found in the vascular endothelium, and the NO produced there plays a key role in vasodilation and vascular remodeling (Rudic et al, 1998). eNOS expression is stimulated by shear stress by activation of the tyrosine kinase c-SRC (Davis et al, 2004), by heat shock protein 90 (Harris et al, 2003), by oxidant stress (Cai et al, 2001), and by vascular mediators such as bradykinin, serotonin, adenosine, ADP/ATP, histamine, and thrombin (Arnal et al, 1999). After its formation by vascular endothelial cells, NO diffuses to vascular smooth muscle cells where it activates soluble guanylyl cyclase (sGC), producing 3′,5′-cyclic guanosine monophosphate (cGMP). Subsequently, cGMP activates both cGMP- and 3′,5′-cyclic adenosine monophosphate (cAMP)-dependent protein kinases (PKG and PKA, respectively) leading to smooth muscle relaxation. eNOS blockade increases renal vascular resistance and decreases the glomerular ultrafiltration coefficient (Gabbai, 2001). NO also helps maintain vascular integrity, with increased expression being linked to decreased neointimal formation and medial thickening (Kawashima et al, 2001). Indeed, the degenerative changes seen in chronic allograft nephropathy related to cyclosporine use can be mitigated by increased nitric oxide expression (Chander et al, 2005). Increased eNOS activity is also associated with protection from renal ischemia-reperfusion injury (Shoskes et al, 1997). Although raised local levels of NO from eNOS can be beneficial to renal function, induction of iNOS and overproduction of NO from inflammatory cells can be deleterious. In the face of free oxygen radicals at the site of inflammation, NO can interact with reactive oxygen species to form peroxynitrite, which induces protein damage by formation of nitrotyrosine. Increased iNOS has been related to damage from nitrotyrosine in glomerular disease (Trachtman, 2004), lupus nephritis (Takeda et al, 2004), and transplant rejection (Albrecht et al, 2002). Increased iNOS activity has direct renal effects as well, including upregulation of sodium and bicarbonate tubular transport (Wang, 2002). Carbon monoxide (CO) gas is another reactive diffusible mediator with multiple effects throughout the body, especially in the kidney. Heme oxygenase (HO), an essential enzyme in heme catabolism, catalyzes the rate-limiting step in heme degradation, resulting in the formation of iron, carbon monoxide, and biliverdin (Hill-Kapturczak et al, 2002). Biliverdin is subsequently converted to bilirubin by biliverdin reductase. HO is expressed in two forms, constitutive HO-2 and inducible HO-1. Increased CO production produces vasodilation in the kidney and can counteract catecholamine induced vasoconstriction (Mustafa and Johns, 2001). In particular, both HO-1 and HO-2 are highly expressed in the medulla and help maintain renal medullary blood flow (Zou et al, 2000). In cirrhosis, decreased renal expression of HO-1 is linked to renal dysfunction (Miyazono et al, 2002). CO also regulates sodium transport in the loop of Henle, with HO-2 blockade inhibiting sodium excretion (Wang et al, 2003) and stimulation increasing natriuresis and diuresis (Rodriguez et al, 2003). The other primary effect of CO in the kidney is renoprotection from oxidant injury. CO has documented anti-inflammatory, antioxidant, and cytoprotective actions (Sikorski et al, 2004). Indeed, a patient with a genetic HO-1 deficiency had significant tubular and vascular endothelial injury (Ohta et al, 2000). Increased CO is protective against ischemia-reperfusion injury in native and transplant kidneys (Nakao et al, 2005). Induction of HO-1 through agents such as bioflavonoids protects against tubular damage and improves renal transplant function (Shoskes et al, 2003). Mature RBCs are produced from a small pool of multipotent progenitor cells (Suda et al, 1984), which in turn are derived from the fetal liver. The earliest committed cell is the erythroid burst-forming unit (BFU-E), which, under appropriate stimulation, divides to produce erythroid colony-forming units (CFU-E). Further differentiation leads to the production of proerythroblasts, reticulocytes, and, ultimately (after extrusion of the nucleus), mature RBCs. The entire process requires about 2 weeks. EPO-deficient mice die in utero with a marked reduction in erythropoiesis (Munugalavadla and Kapur, 2005). Additional growth factors such as interleukin-3, GM-CSF, stem cell factor, activin, insulin-like growth factor (IGF-I), and, possibly, hepatic growth factor act synergistically with EPO to reduce apoptosis and thus promote proliferation of erythroid cells (Muta and Krantz, 1993). EPO has also been shown to have effects outside of the bone marrow. EPO receptors have been demonstrated in kidney, brain, retina, heart, skeletal muscle, and endothelial cells (Juul et al, 1998). In the kidney, pretreatment with high-dose EPO has been shown to reduce ischemia-reperfusion injury in animal models, due to decreased apoptosis (Patel et al, 2004). Production of EPO, and hence erythropoiesis, is closely associated with circulating O2 tension. Under hypoxic conditions, the alpha subunit of the regulatory protein hypoxia-inducible factor-1 (HIF-1) is exposed (Wang et al, 1995). Binding of HIF-1 alpha with HIF-1 beta, hepatic nuclear factor-4 (HNF-4), and p300 turns on erythropoietin transcription (Arany et al, 1996). Once the hypoxia has been corrected, HIF-1 alpha is ubiquinated and rapidly degraded by proteosomes, thus shutting down erythropoietin production. There is also in-vitro evidence that hypoxia itself may directly increase erythropoiesis through HIF-1–mediated increases in autocrine motility factor (AMF) production and subsequent decrease in apoptosis (Mikami et al, 2005). In states of chronic inflammation, erythropoiesis is decreased. Apoptosis of erythroid progenitor cells occurs in the presence of the tumor-associated antigen RCAS1, which is also produced by macrophages under inflammatory conditions (Suehiro et al, 2005). In certain malignancies, such as renal cell carcinoma, erythropoiesis is enhanced due to a mutation in the von Hippel-Lindau (VHL) gene. As a result, there are constitutively increased levels of HIF-1 and polycythemia (Wiesener et al, 2002). rHuEPO has also been used to treat anemia associated with malignancy but must be used with caution in these conditions because its use may be associated with an increased risk of venous thromboembolism and higher mortality (Bennett et al, 2008). Normal regulation of bone mineralization, through maintenance of serum calcium and phosphorus levels, is achieved through the actions of vitamin D and parathyroid hormone (PTH). The actions of both hormones are exerted largely through the kidney (Fig. 38–3). Figure 38–3 Effects of vitamin D and parathyroid hormone (PTH) on calcium homeostasis. ECF, extracellular fluid. (From Yu SLY. Renal transport of calcium, magnesium, and phosphate. In: Brenner BM, editor. Brenner and Rector’s the kidney. 7th ed. Philadelphia: WB Saunders; 2004. p. 536.) The kidney plays an important role in the regulation of vitamin D activity. The major source of vitamin D is through dermal synthesis of the precursor compound cholecalciferol (vitamin D3), or through dietary intake of vitamin D3–fortified foods. Vitamin D3 has minimal biological activity and requires two hydroxylations to become active. The first occurs in the liver through the action of 25-hydroxylase to form 25-hydroxycholecalciferol (calcidiol). The calcidiol molecule is bound to vitamin D binding protein and transported to the kidney, where it is filtered and reabsorbed by renal tubular cells. A second hydroxylation occurs within the tubular cell. Because these cells contain both 1α-hydroxylase and 24α-hydroxylase, hydroxylation will produce either inactive 24,25-dihydroxycholecalciferol or 1,25-dihydroxycholecalciferol (calcitriol), the biologically active form that is one hundred times more potent than calcidiol. Calcitriol production is regulated by calcidiol levels as well as the 1α-hydroxylase levels. These are, in turn, determined by PTH and plasma phosphate levels (increased enzyme activity) and serum calcitriol levels (decreased enzyme activity) (Portale et al, 1989). However, unregulated calcitriol synthesis can occur in macrophages in granulomatous conditions such as sarcoidosis and tuberculosis, and in prostate epithelial and cancer cells (Young et al, 2004). Calcitriol functions through a single intracellular vitamin D receptor (VDR) to regulate gene transcription (Lowe et al, 1992). Its primary function is the maintenance of serum calcium and phosphorus levels. The four main target organs are the intestine (increases intestinal absorption of calcium, and to a lesser extent, phosphorus), the bones (regulates osteoblast activity, and in combination with PTH, allows for osteoclast activation and bone resorption), the kidney (increases reabsorption of calcium) and the parathyroid gland (suppresses PTH release). Recent evidence suggests that both calcidiol and calcitriol may also function as antiproliferative agents. Prostate epithelial and cancer cells demonstrate VDR, and vitamin D may suppress the growth of these cells, especially in combination with androgens (Tuohimaa et al, 2005). The renal effects of PTH are threefold. First, it increases active calcium reabsorption at the level of the distal tubule (Friedman and Gesek, 1993). Second, it decreases phosphate reabsorption in the proximal convoluted tubule (and the distal tubule, to a lesser degree) through its action on the sodium phosphorus cotransporter (Pfister et al, 1997). Third, it stimulates calcitriol production by increasing 1α-hydoxylase levels while decreasing 24α-hydroxylase levels (Broadus et al, 1980). ADH increases the passive reabsorption of water at the level of the collecting duct. Through interaction with the V2 receptor, it facilitates the insertion of preformed water channels, known as aquaporin-2 (AQP-2), into the luminal membrane of the principal cells (Agre et al, 2002). This allows luminal water to enter the cell and then diffuse back into the systemic circulation through the basolateral membrane of the cell (Fig. 38–4). ADH increases urea reabsorption in the medullary collecting tubule through specific urea transporters, which helps maintain the high interstitial osmolality required for water reabsorption. ADH also increases systemic vascular resistance through interaction with the V1 receptor; this is of minor physiologic importance. Other effects of ADH include increased sodium reabsorption and potassium excretion, increased prostaglandin synthesis, increased ACTH secretion (through V3 receptors), and release of both factor VIII and von Willebrand factor from vascular endothelium. (From Brown D, Nielsen S. The cell biology of vasopressin action. In: Brenner BM, editor. Brenner and Rector’s the kidney. 7th ed. Philadelphia: WB Saunders; 2004. p. 574.) There are two major stimuli for ADH release, hyperosmolality and decreased effective circulating volume (ECV), as well as a number of less common factors (Table 38–2). Table 38–2 Physiologic and Pathologic Factors Affecting the Release of Antidiuretic Hormone The major extracellular osmole is sodium; so, for practical purposes ADH release is governed by changes in serum sodium concentrations. Serum osmolality is monitored by osmoreceptors in the hypothalamus, and changes as little as one percent are enough to affect ADH release (Robertson, 1987). There are a number of other factors that can increase ADH secretion (see Table 38–2). Nausea and pain are probably the most clinically relevant, and, as a result, postsurgical hyponatremia due to excessive ADH release is a potentially life-threatening problem. When both decreased ECV and hyponatremia coexist, the pressure receptors usually override the osmoreceptors and prevent the inhibition of ADH secretion that is usually seen with hyponatremia (Baylis, 1987). This is clinically relevant in conditions of decreased ECV and hyponatremia, such as congestive heart failure where ADH secretion persists despite significant hyponatremia. The renal tubule has two basic functions: reabsorption (transport of substances from lumen to blood) and secretion (transport of substances from blood to lumen). Transport can involve one of two pathways: either transcellular (across the luminal and basolateral membrane) or paracellular (between cells) (Fig. 38–5). Each section of the tubule (Fig. 38–6) is specialized to facilitate absorption and secretion of certain substances through a variety of transport mechanisms. Figure 38–5 Example of transcellular transport between the tubule cell and the basolateral membrane. (From Moe OW, Baum M, Berry CA, Rector FC Jr. Renal transport of glucose, amino acids, sodium, chloride, and water. In: Brenner BM, editor. Brenner and Rector’s the kidney. 7th ed. Philadelphia: WB Saunders; 2004. p. 425.) Figure 38–6 Organization of the renal tubule. (From Knepper MA, Gamba F. Urine concentration and dilution. In: Brenner BM, editor. Brenner and Rector’s the kidney. 7th ed. Philadelphia: WB Saunders; 2004. p. 601.) The PCT is responsible for reabsorption of 60% of the glomerular filtrate. Under normal circumstances it reabsorbs 65% of the filtered sodium, potassium, and calcium; 80% of filtered phosphate, water, and bicarbonate; and 100% of the filtered glucose and amino acids (Moe et al, 2004). The PCT is able to increase or decrease reabsorption in response to changes in GFR to maintain constant reabsorptive fractions through the process of glomerulotubular balance. This is accomplished mainly by the early (S1 and S2) segments of the PCT. The later (S3) segment is responsible for secretion of numerous drugs and toxins that are too large, or protein bound, to be filtered. As well, the PCT is responsible for the generation of ammonia from glutamine, which is necessary for urinary acidification. The majority of sodium reabsorption occurs in the PCT, and occurs through both secondary active and passive mechanisms (Fig. 38–7). Figure 38–7 Mechanisms of sodium reabsorption in the proximal tubule. (From Moe OW, Baum M, Berry CA, Rector FC Jr. Renal transport of glucose, amino acids, sodium, chloride, and water. In: Brenner BM, editor. Brenner and Rector’s the kidney. 7th ed. Philadelphia: WB Saunders; 2004. p. 414.)
Renal Physiology
Vascular (Renal Blood Flow and Glomerular Flow Rate)
Renal Blood Flow (RBF)
Determinants of Glomerular Filtration
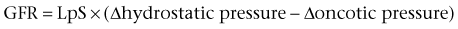
Regulation of Glomerular Filtration Rate
Clinical Assessment of Glomerular Filtration Rate
Renal Clearance
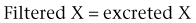
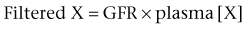
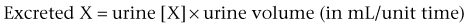
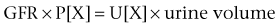

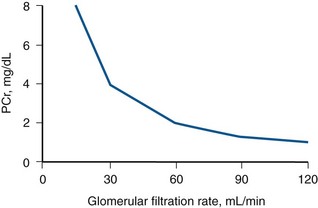
Plasma Markers
Mathematical Correction
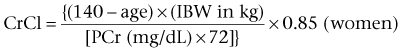
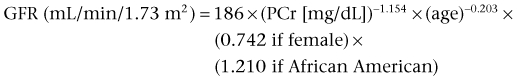
Hormonal
Control of Renal Vascular Tone
Vasoconstriction
Vasodilation
Vasoconstrictors
Angiotensin II
Norepinephrine
Endothelin
Vasopressin
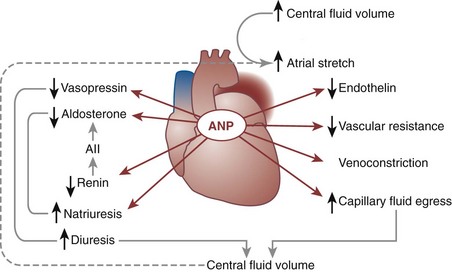
Atrial Natriuretic Peptide
Vasodilators
Nitric Oxide
Carbon Monoxide
Erythropoiesis
Erythroid Progenitor Cells
Erythropoietin
Regulation of EPO Production and Erythropoiesis
Bone Mineral Regulation
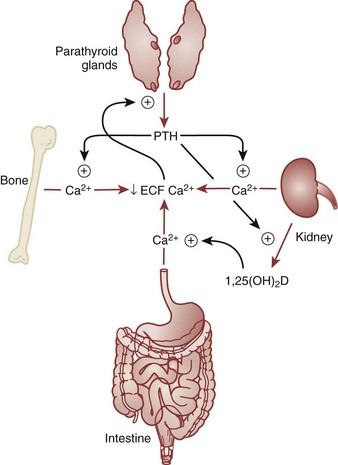
Vitamin D Regulation
Vitamin D Activity
Parathyroid Hormone Activity
Kidney
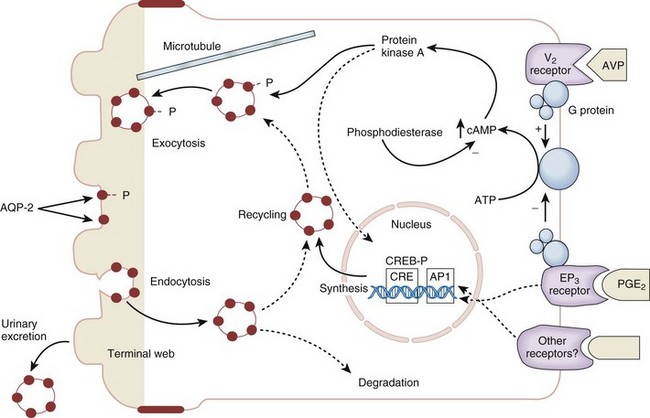
Antidiuretic Hormone
ADH Actions
Control of ADH Secretion
STIMULI
INHIBITORS
Hyperosmolality
Hypo-osmolality
Hypovolemia
Hypervolemia
Stress (e.g., pain)
Ethanol
Nausea
Phenytoin
Pregnancy
Hypoglycemia
Nicotine
Morphine
Other drugs
Hyperosmolality
Other Stimuli
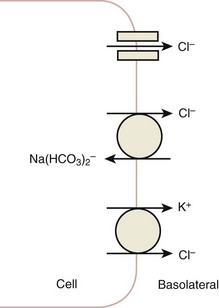
Renal Tubular Function
Basic Functions
Proximal Convoluted Tubule (PCT)
Sodium
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree