have designated this condition as ischemic nephropathy, which some believe is a major cause for some patients reaching end-stage renal disease (ESRD).6,7 More recently, attention has been focused on the role of renovascular disease in impairing cardiac function, both by reducing the systemic excretion of sodium and volume and by producing abrupt rises in arterial afterload that magnify cardiac dysfunction. The primary task of the clinician is to elucidate the role of renal arterial stenosis in a given patient and to direct therapy accordingly.
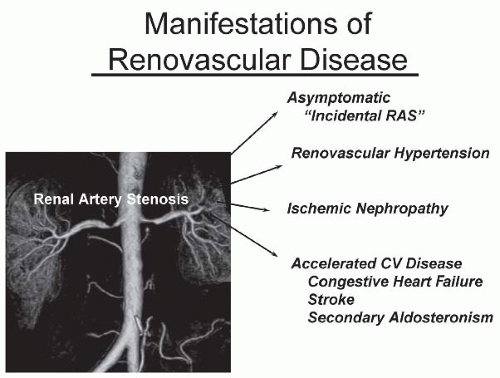 FIGURE 42.1 Clinical manifestations of renal artery stenosis range across a broad spectrum. Many, perhaps most, represent incidental lesions with minimal hemodynamic effects. Some reach levels wherein the activation of pressor mechanisms produces a rise in blood pressure, identified as renovascular hypertension, and at some point, threaten kidney function sufficiently to warrant the term ischemic nephropathy (see text). Particularly when the entire renal mass is affected, impaired kidney function and solute excretion from renovascular disease can accelerate cardiovascular morbidity, sometimes identified as one of the cardiorenal syndromes, with worsening congestive heart failure. The task facing the clinician is to identify where along this spectrum an individual patient lies. CV, cardiovascular; RAS, renal artery stenosis. |
but identified relatively high associated morbidities and mortalities, particularly in patients with atherosclerotic disease.
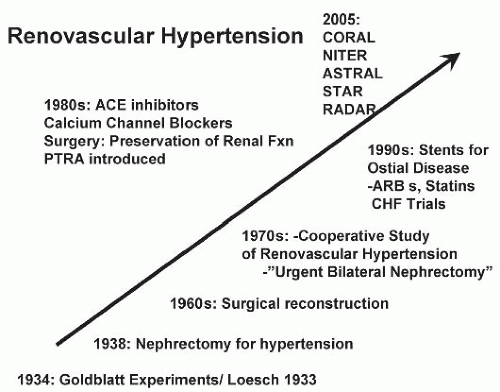 FIGURE 42.2 A time line with major events in the understanding of renovascular hypertension. Goldblatt and Loesch are identified as the original investigators in the 1930s who linked reduced kidney perfusion to the development of sustained hypertension. Surgical revascularization only became technically feasible in the 1960s, with the emergence of effective medical therapy and endovascular procedures in the 1980s and 1990s. The application of effective medical therapy, including statins, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin-receptor blockers (ARBs), have led to clinical equipoise that is the basis for prospective, randomized trials comparing optimal medical treatments with or without revascularization, beginning around 2005. PTRA, percutaneous transluminal renal angioplasty; CORAL, cardiovascular outcomes for renal atherosclerotic lesions; NITER, nephrotherapy ischemic therapy; ASTRAL, angioplasty and stent for renal artery lesions; STAR, stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function; RADAR, randomized, multicenter, prospective study comparing best medical treatment versus best medical trearment plus stenting in patients with hemodynamically relevant atherosclerotic renal artery stenosis; CHF, congestive heart failure. |
Atherosclerotic renal artery disease is the most common cause of renal artery stenosis, accounting for about 80% of renal arterial lesions. Fibrous renal artery diseases, as a group, account for less than 20% of renal arterial lesions. Reports of resistant hypertension suggest that when renovascular disease is a factor, more than 84% is atherosclerotic renal artery disease and 16% is fibromuscular renal artery disease.13 Fibrous renal artery disease has been reported in 2% to 6% of potential renal transplant donors (usually normotensive individuals).14,15 The vast majority of incidental renal artery lesions in angiographic series are atherosclerotic.
TABLE 42.1 Lesions Producing Renovascular Hypertension and Ischemic Nephropathy | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
TABLE 42.2 Histologic Classifications of Fibromuscular Dysplasia and Angiographic Appearance | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
in bands, sometimes with disruption of the vascular elastic membrane. The molecular basis for these disruptions is unknown, although several candidate genes have been proposed. The primary subtypes of FMDs are designated as medial fibroplasia, intimal fibroplasia, and periadventitial fibroplasia, defined initially on the basis of histologic analysis of surgically resected vessels. Medial fibroplasia is the most common phenotype, and often manifests as a string-of-beads appearance with alternating stenoses with apparent luminal dilations, as summarized in Table 42.2. The other phenotypes appear as focal or elongated vascular occlusions. Medial fibroplasia affects the distal half of the main renal artery, frequently extends into the major branches, is often bilateral, and angiographically gives the appearance of multiple aneurysms wherein the diameters of the aneurysms are wider than the apparently unaffected portion of the main renal artery (Fig. 42.3A). Most cases of medial fibroplasia are diagnosed in women between the ages of 30 and 50 years. Although medial fibroplasia progresses to higher degrees of stenosis in about one-third of patients, complete arterial occlusion and/or ischemic atrophy of the kidney ipsilateral to the renal artery stenosis are rare. The stenotic lesions in medial fibroplasia are secondary to thickened fibromuscular ridges replacing the normal structure of the intima and the media of the artery. These thickened ridges alternate with thinned areas that may not have an internal elastic membrane, thereby becoming aneurysmal.
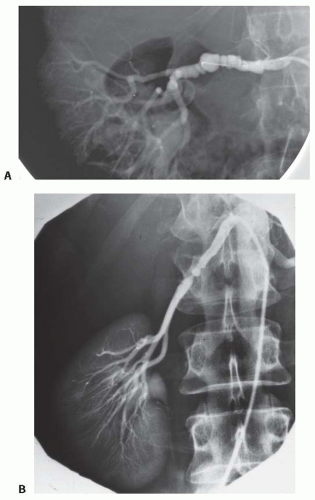 FIGURE 42.3 Examples of fibromuscular disease of the renal artery. A: An image of the string of beads appearance that is typical of medial fibroplasia with aneurysmal bulging beyond the artery. B: An image of a tight, focal lesion that is typical of intimal hyperplasia in a young male. |
and its main branches, including the common carotid, and the subclavian and the renal arteries.22 Renal artery involvement is not uncommon in Asian and African patients and is a major cause of renovascular hypertension in these countries.23 Koide24 reported renovascular hypertension in 278 (18.8%) of 1,475 patients in a Japanese nationwide survey of Takayasu arteritis. Takayasu arteritis is a common cause of renovascular hypertension in Southeast Asia, including India and China, and accounts for between 20% and 60% of RVH cases.23,25
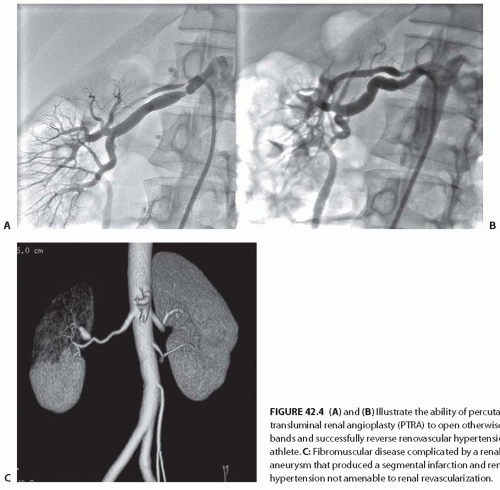 FIGURE 42.4 (A) and (B) Illustrate the ability of percutaneous transluminal renal angioplasty (PTRA) to open otherwise tight fibrous bands and successfully reverse renovascular hypertension in a young athlete. C: Fibromuscular disease complicated by a renal artery aneurysm that produced a segmental infarction and renovascular hypertension not amenable to renal revascularization. |
cause of RVH and can contribute to a loss of renal function leading to ESRD (Fig. 42.5B,C). Atherosclerotic plaque often arises in the first 1 to 2 cm of the renal artery or may extend directly from the aorta into the renal ostium. Aortic and renal vascular calcification is often present. Some 75% to 80% of patients with renal artery atherosclerosis and renovascular hypertension have ostial atherosclerotic lesions, and 25% to 30% of lesions are in the nonostial location. ARAS is a manifestation of systemic atherosclerotic disease and is asso ciated with coronary, cerebrovascular, peripheral vascular, and aortic disease.29,30 The prevalence of ARAS appears to be increasing. This probably reflects the fact that more people are living long enough for atherosclerotic vascular disease in the visceral abdominal vessels to reach critical levels, thus aggravating hypertension when the kidney is affected. In a recent systematic analysis of patients undergoing an angiography of the peripheral or coronary circulations, ARAS was found in 11% to 42% of cases.30 Predictors of ARAS include a history of hypertension, the presence of renal functional impairment, coexisting vascular or coronary artery disease, the presence of abdominal bruits, and a history of smoking. Renal artery lesions are bilateral in 20% to 40% of such patients. Estimates of the prevalence of ARAS depend on the population screened. One population-based study of a cohort of 870 patients older than 65 years screened with renal artery duplex sonography found a 6.8% prevalence of ARAS, defined as greater than 60% stenosis. No differences in prevalence were detected between African Americans and Caucasians,31 although reported rates for surgically corrected renovascular hypertension are lower for African Americans.32 Men are affected more often than women, but this gender difference declines with advancing age. Recent series indicate a distinct shift toward women in series referred for revascularization.33,34 Several studies suggest that atherosclerotic renal vascular disease is associated with adverse coronary events33,35 and increased mortality. Autopsy series report an overall prevalence of 4% to 20%, with progressively higher rates for those older than 60 years (25% to 30%) and 75 years (40% to 60%). These studies suggest that ARAS leading to renal artery stenosis is the single most common cause of secondary hypertension in patients older than 50 years.36 It also commonly leads to systolic hypertension with wide pulse pressures. Furthermore, renal artery stenosis has been reported to contribute
to the decline in renal function in 15% to 22% of patients reaching ESRD.37,38
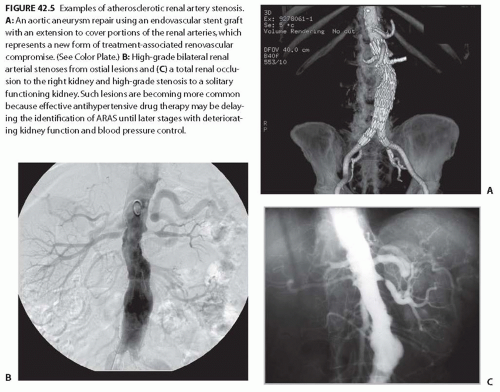 FIGURE 42.5 Examples of atherosclerotic renal artery stenosis. A: An aortic aneurysm repair using an endovascular stent graft with an extension to cover portions of the renal arteries, which represents a new form of treatment-associated renovascular compromise. (See Color Plate.) B: High-grade bilateral renal arterial stenoses from ostial lesions and (C) a total renal occlusion to the right kidney and high-grade stenosis to a solitary functioning kidney. Such lesions are becoming more common because effective antihypertensive drug therapy may be delaying the identification of ARAS until later stages with deteriorating kidney function and blood pressure control. |
perfusion pressures. Luminal occlusion of less than 60% (cross-sectional area) rarely produces any measurable gradient for either pressure or flow. Hence, a fall in renal perfusion pressure sufficient to initiate RVH occurs only when luminal occlusion is relatively severe, usually in the 70% to 80% cross-sectional occlusion range (Fig. 42.7). When critical stenosis develops and reduces renal perfusion pressure, multiple mechanisms are activated in the kidney to restore renal blood flow. Foremost among these pathways is the release of renin from the juxtaglomerular apparatus, leading to the activation of the renin-angiotensin-aldosterone system (RAAS). Release of plasma renin occurs only after poststenotic pressures fall by at least 10% to 20% compared with aortic pressures.43 This is mediated in part by the stimulation of neuronal nitric oxide synthase and cyclooxygenase 2 in the macula densa. Blockade of the RAAS at the time an experimental renal artery lesion is created prevents the development of hypertension. Animals genetically modified to lack the angiotensin (Ang 1) receptor fail to develop two-kidney one-clip hypertension.44 Experiments using kidney transplantation from AT1 receptor knockout mice indicate that both systemic and renal angiotensin receptors participate in additive fashion to blood pressure regulation.45
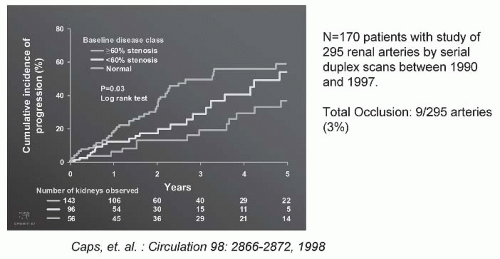 FIGURE 42.6 The progression of atherosclerotic renal artery stenosis (ARAS) of varying severity during a serial follow-up by Doppler ultrasound. There were 295 arteries that were followed sequentially at 6-month intervals for 5 years. Measurable hemodynamic progression (defined as an increase in peak systolic velocity of at least 100 cm per second) was identified in 31% by 3 years, and more than 50% of the group with more than 60% at baseline. Importantly, clinical progression was uncommon (defined by a change in serum creatinine, loss of kidney size, or total occlusion). Predictors of progression included systolic blood pressure, age, and diabetes. (Adapted from Caps MT, Perissinotto C, Zierler RE, et al. Prospective study of atherosclerotic disease progression in the renal artery. Circulation. 1998;98:2866-2872.) (See Color Plate.) |
beyond a stenosis. There is no normal or nonstenotic kidney to counteract increased systemic pressures. As a result, sodium is retained and the blood volume is expanded, which eventually feeds back to inhibit the renin-angiotensin system (RAS) (Fig. 42.8B). Therefore, 1K-1C hypertension is typically not angiotensin dependent unless the removal of volume is achieved that reduces renal perfusion pressure and again activates the RAAS.
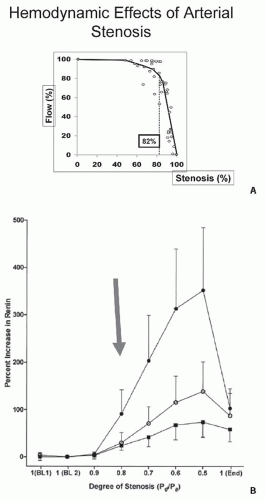 FIGURE 42.7 A: Arterial pressure depicted as a function of degree of luminal occlusion. These data highlight the fact that no change in postlesion pressure or flow can be identified until cross-sectional occlusion is severe, usually more than 70% to 80%. These experimental data in dogs are consistent with measurements in human subjects (B), wherein renal vein renin release was not detected during balloon occlusion until translesional gradients between the aorta and renal artery of at least 10% to 20% were produced. MAP, mean arterial pressure. (Data from May et al.229 and De Bruyne et al.43) |
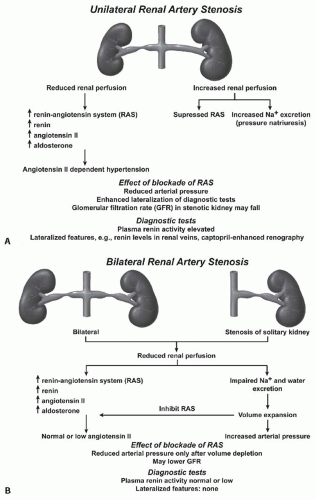 FIGURE 42.8 A: A schematic illustration of hormonal and hemodynamic effects of unilateral renal artery stenosis to produce renovascular hypertension. Unilateral disease is identified as 1-clip-2-kidney Goldblatt hypertension and manifests with the activation of the renin-angiotensin system so long as the contralateral kidney continues to undergo pressure natriuresis (see text). (Adapted from Safian and Textor91 .) Panel (B) illustrates the sequence of events with 1-clip-1-kidney Goldblatt hypertension, wherein no normal contralateral kidney is available to excrete sodium and volume in response to rising arterial pressures. In this model, the initial activation of the renin-angiotensin system is temporary due to volume expansion. |
kidney sodium reabsorption) may be problematic, because high levels of Ang II and aldosterone stimulate sodium reabsorption in both the stenotic and nonstenotic kidney.
extracellular fluid volume expansion persists. In phase 3 of 2K-1C hypertension, acute blockade of the RAS fails to lower the BP. Sodium depletion may ameliorate the hypertension, but does not normalize it.
TABLE 42.3 Interactive Mechanisms Underlying Hypertension and Kidney Injury in Atherosclerotic Renal Artery Stenosis | ||||||
|---|---|---|---|---|---|---|
| ||||||
microcirculation may explain some of the limitations observed after the restoration of large-vessel patency (e.g., with renal revascularization). Occasionally, renovascular disease is associated with nephrotic range proteinuria that can regress after renal revascularization.76 The mechanism of enhanced glomerular permeability in this context is unknown.
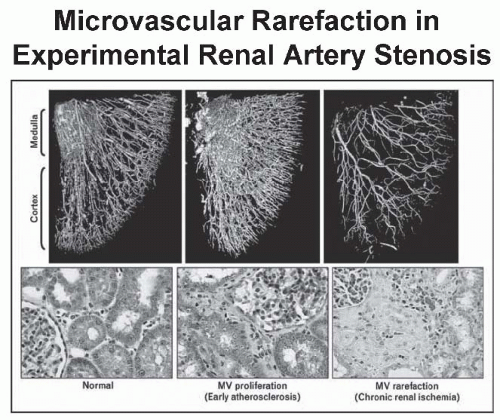 FIGURE 42.9 Reconstructions of the vascular structures using micro-computed tomography (CT) imaging in experimental renal artery stenosis. The atherosclerotic milieu produced by cholesterol feeding leads to microvascular (MV) proliferation and renders the animal especially susceptible to rarefication and obliteration of microvessels in the setting of high-grade renal artery stenosis. These changes eventually lead to interstitial fibrosis and a loss of kidney function. (From Lerman LO, Chade AR. Atherosclerotic process, renovascular disease and outcomes from bench to bedside. Curr Opin Nephrol Hyper. 2006;15:583-587, with permission.) |
proteinuria, which can regress with the correction of the vascular lesions.
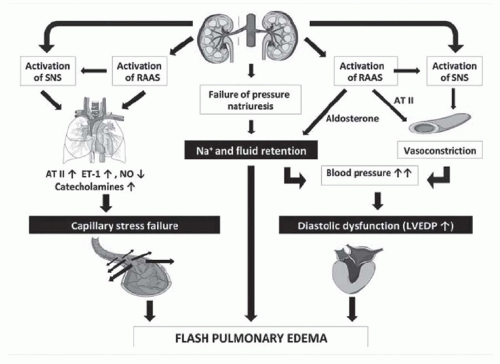 FIGURE 42.10 A schematic depicting the interplay of mechanisms leading to rapidly developing symptoms of left ventricular heart failure sometimes observed with renal artery stenosis. These include impaired volume excretion with bilateral atherosclerotic renal artery stenosis (ARAS), a rise in afterload and impaired left ventricular pump function, as well as disturbed capillary function within the pulmonary vasculature in part related to the activation of neurogenic and hormonal pathways. SNS, sympathetic nervous system; RAAS, renin-angiotensin-aldosterone system; AT II, angiotensin II; ET-1, endothelin 1; NO, nitric oxide; LVEDP, left ventricular end-diastolic pressure. (From Messerli FH, Bangalore S, Makani H, et al. Flash pulmonary oedema and bilateral renal artery stenosis: the Pickering syndrome. Eur Heart J. 2011;32(18):2231-2237, with permission.) |
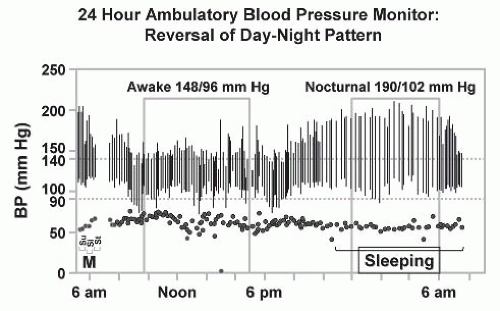 FIGURE 42.11 Ambulatory blood pressure (BP) monitoring in patients with renovascular hypertension commonly reveals a loss of nocturnal pressure fall, or in some cases, a complete reversal of the day-night pattern as shown here. This may be one of the features by which target-organ injury such as left ventricular hypertrophy is exaggerated as compared to patients with similar daytime blood pressure levels.232 |
When combined with the ambiguous results of prospective treatment trials in which medically treated individuals with ARAS fared as well as those treated with renal revascularization, some would argue that there is little evidence to support extensive diagnostic studies to identify all cases of renovascular hypertension. As a result, most patients with hypertension simply are treated and subjected to few laboratory investigations. For those who reach acceptable BP control without adverse effects, no further studies are performed. Hence, many if not most cases of true RVH are not detected (Fig. 42.12) unless hypertension becomes more difficult to treat or if renal dysfunction ensues. One important reason that RVH is less frequently detected is the availability of orally active antihypertensive agents that block the RAAS. Early studies beginning with captopril indicated that satisfactory BP control can be achieved in more than 86% of patients with RVH compared with less than 50% with previously available drugs. In recent years, the widespread application of ACE inhibitors and angiotensin receptor blockers (ARBs) for indications other than hypertension (e.g., congestive cardiac failure, diabetic nephropathy, and other proteinuric renal disease) increases the exposure of individuals with undetected renal artery stenosis to these drugs.
TABLE 42.4 Clinical Features Suggestive of Renovascular Disease | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
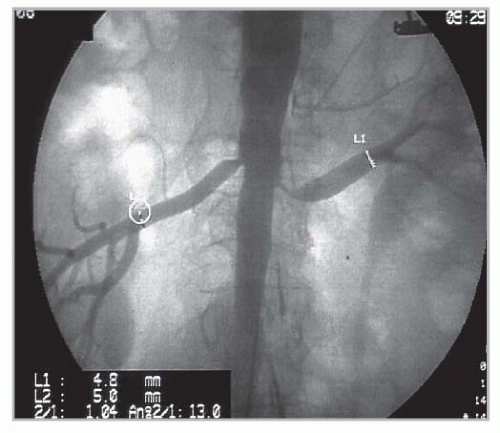 FIGURE 42.12 An aortogram revealing bilateral renal arterial disease performed during a coronary angiography. This individual was treated with antihypertensive drug therapy with excellent blood pressure (BP) control and normal kidney function and was identified only because imaging was included with coronary studies. Numerous series indicate that many such patients are otherwise never identified, but can be managed with medical therapy only. The number that progress to more advanced disease and/or loss of kidney function with current medical therapy may be less than 10% (see text). |
presence of bilateral renal artery stenosis, or stenosis in a solitary functioning kidney.84,85,86,87,88 In one series, more than half of patients demonstrating an acute elevation in the plasma creatinine concentration that was either unexplained or occurred shortly after the institution of therapy with an ACE inhibitor had main renal artery disease, whereas the remainder presumably had a disease of the intrarenal vessels due to nephrosclerosis.88 Remarkably, most patients with renal artery stenosis tolerate ACE inhibition or ARB Rx with few adverse effects.89
TABLE 42.5 Goals of Diagnostic and Therapeutic Intervention in Renovascular Hypertension and Ischemic Nephropathy | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
be due to estrogen intake, pregnancy, cortisol excess, intrarenal microvascular ischemia (parenchymal disease), or accelerated or malignant hypertension, and the 15% to 20% of patients with primary (essential) hypertension and high renin levels constitute the majority of high-renin hypertensives. Renin secretion fluctuates widely and is influenced by sodium intake, posture and sympathetic tone,96 a variety of drugs, age, sex, and race.97 The utility of peripheral PRA is reportedly enhanced when measured in the morning with the patient in the seated position and when indexed against urinary sodium excretion; when measured under these exacting circumstances, a high peripheral PRA is found in 75% to 80% of patients with proven renovascular hypertension.98 Other investigators, measuring peripheral PRA under similar circumstances, failed to demonstrate significantly increased sensitivity of the peripheral PRA in predicting either the presence of ARAS and the response to therapy.92,99,100
TABLE 42.6 Noninvasive Assessment of Renal Artery Stenosis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


