Pierre M. Ronco, Pierre Aucouturier, Bruno Moulin
Renal Amyloidosis and Glomerular Diseases with Monoclonal Immunoglobulin Deposition
The glomerular capillaries are a favorite site for the deposition of abnormal proteins. In most patients, the resulting diseases are caused by a monoclonal immunoglobulin or subunit thereof and can be classified into two categories by electron microscopy (Table 27-1). The first category includes diseases with fibril formation, mainly amyloidosis, and diseases with microtubule formation, including cryoglobulinemic glomerulonephritis (see Chapter 21) and immunotactoid glomerulonephritis. The second disease category is characterized by nonorganized electron-dense granular deposits. These deposits are localized along basement membranes in most tissues, especially in the kidney, and define a disease now termed monoclonal immunoglobulin deposition disease (MIDD). In some cases, monotypic immune complex–like deposits are observed in the setting of proliferative glomerulonephritis.
Table 27-1
Glomerular diseases with tissue deposition or precipitation of monoclonal immunoglobulin components.
| Glomerular Diseases with Tissue Deposition or Precipitation of Monoclonal Immunoglobulin Components | |
| Immunoglobulin Deposits | Glomerular Disease |
| Organized | |
| Fibrillar | Amyloidosis (AL, AH) |
| Microtubular | Cryoglobulinemia; immunotactoid glomerulonephritis |
| Nonorganized: Granular | |
| Monoclonal immunoglobulin deposition disease: light-chain, heavy-chain, and light-plus-heavy-chain deposition diseases Immune complex–like proliferative glomerulonephritis | |
Renal Amyloidosis
General Characteristics of Amyloidosis
Definition
Amyloidosis is a generic term for a family of diseases defined by morphologic criteria. The diseases are characterized by the deposition in extracellular spaces of a proteinaceous material. Amyloid deposits are composed of a felt-like array of 7.5- to 10-nm-wide rigid, linear, nonbranching, aggregated fibrils of indefinite length.1 One amyloid fibril is made of two twisted 3-nm-wide filaments, each displaying the typical “cross-β” structure,1 where antiparallel β-sheets are perpendicular to the filament axis.
Amyloid Precursor–Based Classification
Amyloidoses are classified by the type of precursor protein that composes the main component of fibrils2 (Table 27-2). The amyloidogenic propensity is related to the ability of this precursor to form intermolecular β-sheets. Besides these structural properties, which may relate to genetically transmitted mutations, the amyloidogenic potential is enhanced by overproduction or impaired clearance of the precursor.
Table 27-2
Classification of amyloidoses.
The following proteins may also cause amyloidosis: calcitonin, islet-amyloid polypeptides, atrial natriuretic factor, prolactin, insulin, lactadherin, keratoepithelin, and Danish amyloid protein (which comes from the same gene as ABri and has an identical N-terminal sequence). C JD, Creutzfeldt-Jakob disease; FAP, familial amyloidotic polyneuropathy; FCU, familial cold urticaria; FFI, fatal familial insomnia; FMF, familial Mediterranean fever; GSSD, Gerstmann-Sträussler-Scheinker disease; HIDS, hyper-IgD syndrome; MWS, Muckle-Wells syndrome; TRAPS, tumor necrosis factor receptor–associated periodic syndrome.
| Classification of Amyloidoses | ||||
| Amyloid Protein* | Precursor | Distribution | Type | Syndrome or Main Involved Tissues |
| AA | Serum amyloid A | Systemic | Acquired | Secondary amyloidosis, reactive to chronic infection or inflammation, including hereditary periodic fever (FMF, TRAPS, HIDS, FCU, and MWS) |
| AApoAI | Apolipoprotein A-I | Systemic | Hereditary | Liver, kidney, heart, skin, larynx |
| AApoAII | Apolipoprotein A-II | Systemic | Hereditary | Kidney, liver, adrenal glands, spleen, skin |
| Aβ | Aβ protein precursor | Localized | Acquired | Sporadic Alzheimer disease, aging |
| Localized | Hereditary | Prototypic hereditary cerebral amyloid angiopathy, Dutch type | ||
| Aβ2M | β2-Microglobulin | Systemic | Acquired | Chronic hemodialysis |
| ABri | Abri protein precursor | Localized or systemic? | Hereditary | British familial dementia |
| ACys | Cystatin C | Systemic | Hereditary | Icelandic hereditary cerebral amyloid angiopathy |
| AFib | Fibrinogen Aα chain | Systemic | Hereditary | Kidney |
| AGel | Gelsolin | Systemic | Hereditary | Finnish hereditary amyloidosis |
| AH | Immunoglobulin heavy chain | Systemic or localized | Acquired | Primary amyloidosis, myeloma associated |
| AL | Immunoglobulin light chain | Systemic or localized | Acquired | Primary amyloidosis, myeloma associated |
| ALECT2 | Leukocyte chemotactic factor 2 | Systemic | Acquired? | Kidney, liver, adrenal glands |
| ALys | Lysozyme | Systemic | Hereditary | Kidney, liver, spleen, adrenal glands |
| APrP | Prion protein | Localized | Acquired | Sporadic (iatrogenic CJD, new variant CJD) (alimentary?) |
| Localized | Hereditary | Familial CJD, GSSD, FFI | ||
| ATTR | Transthyretin | Systemic | Acquired | Senile heart, vessels |
| Hereditary | Prototypical FAP | |||
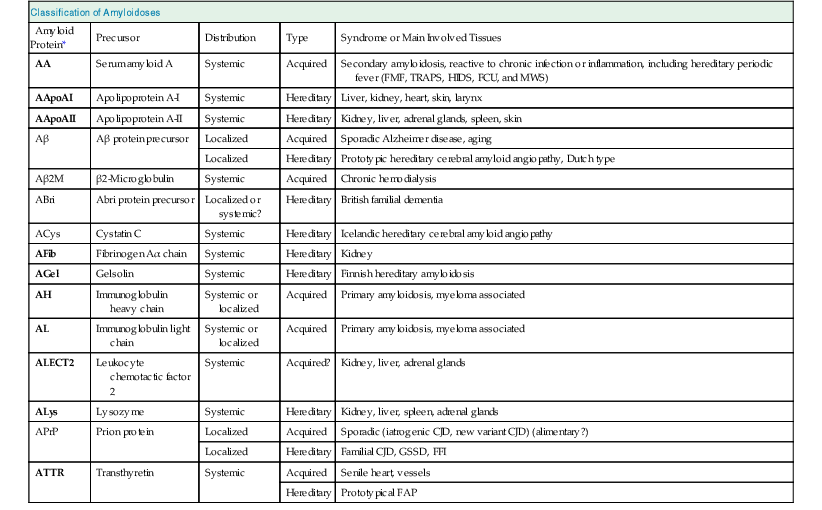
* Entries in boldface indicate amyloid types with kidney involvement.
(Modified from reference 2.)
Renal amyloidoses mostly include immunoglobulin light-chain (AL) and systemic secondary (AA) amyloidoses. Other precursors, such as transthyretin, fibrinogen, apolipoprotein A-I, and lysozyme, are responsible for rare familial cases.
Other Components of All Amyloid Fibrils
Glycosaminoglycans (GAGs) are tightly associated with amyloid fibrils. GAGs are polysaccharide chains made of repeating hyaluronic acid–hexosamine units normally linked to a protein core, thus forming proteoglycans. Proteoglycans, mostly of the heparan sulfate type, appear to induce and to stabilize the β-pleated amyloid structure.
Another constituent of all amyloid deposits is serum amyloid P component (SAP). SAP is resistant to proteolytic digestion, and coating of amyloid fibrils with SAP could result in their protection from catabolism. The high affinity of SAP toward amyloid was exploited for scintigraphy with [123I]-SAP. CPHPC (R-1-[6-[R-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl] pyrrolidine-2-carboxylic acid) is a proline-derived small compound that specifically binds to SAP and allows rapid decreases in serum SAP levels.3 The combination of CPHPC with an SAP-antibody targets amyloid deposits and enables their elimination by recruiting phagocytic cells in a mouse model of AA amyloidosis. A clinical trial using CPHPC combined with an anti-SAP antibody is underway.4
General Mechanisms of Fibrillogenesis
Amyloidogenesis involves a nucleation-dependent polymerization process. Formation of an ordered nucleus is the initial and thermodynamically limiting step, followed by addition of monomers and elongation of the fibrils.5 Fibrillogenesis may involve several mechanisms of processing of the amyloid precursor, including partial proteolysis and conformational modifications. Conformational changes lead to a soluble, partially folded intermediate, whose subsequent ordered self-assembly results in fibril formation. Macrophages have a central role in AA amyloidosis by providing the intralysosomal processing of the precursor. In AL amyloidosis, the variable domain of the light chain VL is the main component, which suggests a role of partial proteolysis of the light-chain precursor.
Pathology
On light microscopy, the deposits are extracellular, eosinophilic, and metachromatic, inducing a change in color of dyes (e.g., crystal violet). After Congo red staining, the deposits appear faintly red (Fig. 27-1, A) and show characteristic apple-green birefringence under polarized light (Fig. 27-1, B). Metachromasia is also observed with crystal violet, which stains the deposits red.
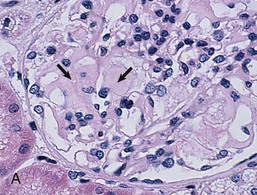
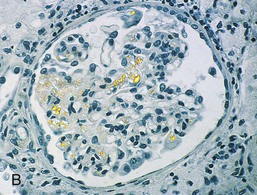
The earliest lesions are located in the mesangium (Fig. 27-1, A), along the glomerular basement membrane (GBM), and in the blood vessels. Mesangial deposits are primarily in the mesangial matrix and spread from lobule to lobule until eventually the whole mesangial area is replaced. Amyloid deposits may also infiltrate the GBM or may be localized on both its sides. When subepithelial deposits predominate, spikes similar to those seen in membranous nephropathy may be observed. Advanced amyloid typically produces a nonproliferative, noninflammatory glomerulopathy and marked enlargement of the kidney. The amyloid deposits replace the normal glomerular architecture, resulting in loss of cellularity. When glomeruli become massively sclerotic, the deposits may be difficult to demonstrate by Congo red staining. In this case, electron microscopy (EM) may be helpful, as well as in the very early stages, which may not be detected by light microscopy examination in patients presenting with the nephrotic syndrome. Amyloid deposits are characterized by randomly oriented, nonbranching fibrils with an 8- to 15-nm diameter (Fig. 27-2).
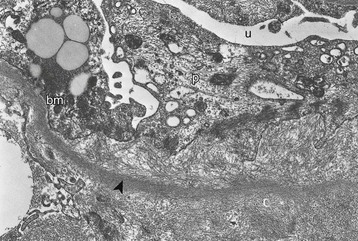
Except for fibrinogen amyloidosis, which characteristically does not affect renal vessels, the media of the blood vessels is prominently involved at early stages. Vascular involvement may predominate and occasionally occurs alone, particularly in AL amyloidosis. Deposits may also affect the tubules and the interstitium, leading to atrophy and disappearance of the tubular structures and to interstitial fibrosis.
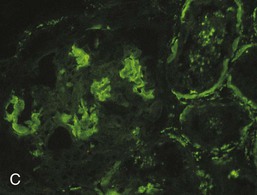
Given the heterogeneity of amyloidoses, immunohistology should be routinely performed (Fig. 27-1, C). Immunohistochemical classification of amyloid type is possible in most cases. Immunohistology with antibodies specific for immunoglobulin chains may be more difficult to interpret than that with anti-AA antiserum, perhaps because of the absence or inaccessibility of light-chain epitopes. Alternative techniques such as immuno-EM6 and mass spectrometry–based proteomic analysis of deposits after laser microdissection, which are still used only at highly specialized centers, will likely decrease the percentage of nontypeable amyloidosis.7,8 A genetic cause should be sought by DNA sequencing in all patients with amyloidosis in whom confirmation of the amyloid precursor cannot be obtained by other techniques.9
Immunoglobulin-Associated Amyloidosis (AL Amyloidosis)
Free immunoglobulin subunits, mostly light chains, secreted by a single clone of B cells, are the cause of the most frequent and severe amyloidosis affecting the kidney. Studies on the mechanisms of AL amyloidogenesis are made particularly difficult by the unique structural heterogeneity of the precursor: each monoclonal light chain is different from all others, so each patient is unique. The involvement of an immunoglobulin heavy chain in amyloidosis (AH and AHL amyloidosis) remains extremely rare, and its diagnosis is greatly enhanced by the use of laser microdissection and mass spectrometry.10
Pathogenesis
Determinant factors are borne by the precursor light chain. In AL amyloidosis, or primary amyloidosis, there is a striking overrepresentation of the lambda (λ) isotype, which is twofold to fourfold more common than the kappa (κ) isotype. A rarely expressed homology family of light-chain variable regions, the VλVI variability subgroup, is found only in amyloid-associated monoclonal immunoglobulins.
Amyloidogenicity is associated with physicochemical features that include low-molecular-mass light-chain fragments in the urine, abnormal disulfide bonding of light chains, and low isoelectric point (pI). An analysis of almost 200 light-chain sequences identified 12 positions in κ chains and 12 in λ chains where certain residues were associated with amyloidosis. Four structural risk factors were shown to define most fibril-forming κ light chains.11 Because of their high dimerization constants, light chains from patients with AL amyloidosis may behave as antibodies with affinity for extracellular structures that could favor a nucleation process.
The tropism of organ involvement may be influenced both by the germ-line gene used for the light-chain variable region (VL) and by somatic mutations occurring in the secreting clone.12 Patients expressing a monoclonal light chain of the VλVI subgroup are more likely to present with dominant renal involvement and less frequent cardiac and multisystem disease.13 Patients with κ light chains are more likely to have dominant hepatic involvement. In addition, organ-specific environmental factors are also involved. For example, high intrarenal concentrations of urea enhance fibril formation by reducing the nucleation lag time.
Amyloid light chains may contribute directly to the pathogenesis, independent of extracellular fibril deposition. In the heart and the kidney at least, the infiltration alone does not correlate well with clinical manifestations. Light chains from amyloid patients incubated with mesangial cells induce a macrophage-like phenotype, whereas those from light-chain deposition disease patients induce a myofibroblast-like phenotype.14
Epidemiology
The incidence of AL amyloidosis is nine per 1 million population per year. Fewer than one in four patients with AL amyloidosis is considered to have an overt immunoproliferative disease, which usually is multiple myeloma, although other forms are seen, such as Waldenström macroglobulinemia. Amyloid deposits are found in approximately 10% of all patients with myeloma and in 20% of those with pure light-chain myeloma. The apparent prevalence of myeloma depends on the diagnostic criteria used. Epidemiologic characteristics of primary amyloidosis, that is, amyloidosis without overt immunoproliferative disease, and myeloma are not significantly different. The median age at diagnosis is 64 years in patients with primary amyloidosis, with a slight predominance of male patients, and about 10% of patients are less than 50 years old.15
Clinical Manifestations
The main clinical symptoms at presentation are weakness and weight loss (Table 27-3). Except for bone pain, the initial symptoms in patients with and without myeloma are similar. However, nephrotic syndrome, orthostatic hypotension, and peripheral neuropathy are more frequent in patients with AL amyloidosis without myeloma.16 Amyloidosis is also different from many types of kidney disease in that the kidneys are often enlarged and hypertension is absent even when renal function is impaired. Proteinuria, mainly albuminuria, occurs in the absence of microhematuria. Indeed, the presence of hematuria should prompt examination for a bleeding lesion in the urinary tract. Renal manifestations may also include renal tubular acidosis (mostly as a part of Fanconi syndrome; see Chapter 50) and polyuria-polydipsia (resulting from urinary concentration defect), when amyloid deposits occur around proximal tubules and Henle loops (or collecting ducts), respectively.
Table 27-3
Clinical and laboratory features at presentation in 474 patients with proven light-chain (AL) amyloidosis.
| Clinical Presentation in 474 Patients with Proven AL Amyloidosis | |
| Features | Percentage |
| Initial Symptoms | |
| Fatigue | 62 |
| Weight loss | 52 |
| Pain | 5 |
| Purpura | 15 |
| Gross bleeding | 3 |
| Physical Findings | |
| Hepatomegaly | 24 |
| Palpable spleen | 5 |
| Lymphadenopathy | 3 |
| Macroglossia | 9 |
| Laboratory Findings | |
| Increased plasma cells (bone marrow >6%) | 56* |
| Anemia (hemoglobin <10 g/dl) | 11 |
| Elevated serum creatinine (1.3 mg/dl) (>113 µmol/l) | 45 |
| Elevated alkaline phosphatase | 26 |
| Hypercalcemia (>11 mg/dl) (>2.75 mmol/l) | 2 |
| Proteinuria (>1.0 g/24 h) | 55 |
| Urine light chain | 73† |
| κ chain | 23 |
| λ chain | 50 |
* 15% of patients having a myeloma.
† Of 429 patients.
(From reference 15.)
AL amyloidosis may infiltrate almost any organ other than the brain and therefore can be responsible for a wide variety of clinical manifestations. Restrictive cardiomyopathy is found at presentation in up to one third of patients and causes death in about one half. Infiltration of the ventricular walls and the septum may be recognized by echocardiography. Amyloid may also induce arrhythmias and the sick sinus syndrome. Amyloid deposits in the coronary arteries may result in angina and myocardial infarction. Cardiac troponins and N-terminal pro–brain natriuretic peptide (NT-proBNP) are sensitive markers of myocardial dysfunction and powerful predictors of overall survival in patients with AL amyloidosis.
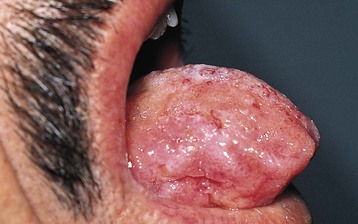
Involvement of the gastrointestinal (GI) tract is common and can cause motility disturbances, malabsorption, hemorrhage, or obstruction. Macroglossia may interfere with eating and obstruct airways (Fig. 27-3). Abnormalities of hepatic function are usually mild. Hyposplenism, diagnosed by abnormal peripheral smear and liver spleen scan and often associated with splenomegaly, predisposes to fatal bacterial infections. Peripheral nerve involvement may result in a painful sensory polyneuropathy, followed later by motor deficits. Autonomic neuropathy causing orthostatic hypotension, lack of sweating, GI disturbances, bladder dysfunction, and impotence may occur alone or together with peripheral neuropathy. Orthostatic hypotension is one of the major hampering complications of AL amyloidosis, with some patients being bedridden. Skin involvement may take the form of purpura, characteristically around the eyes (Fig. 27-4), and ecchymoses, papules, nodules, and plaques, occurring usually on the face and upper trunk. AL amyloidosis may also infiltrate articular structures and mimic rheumatoid or an asymmetric seronegative synovitis. Infiltration of the shoulders may produce severe pain and swelling (shoulder pad sign).
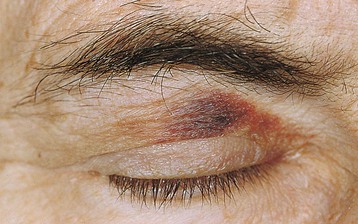
A rare but potentially serious manifestation of AL amyloidosis is an acquired bleeding diathesis that may be associated with deficiency of factor X or factor IX or with increased fibrinolysis. It should be systematically sought before any biopsy of a deep organ. Widespread vascular deposits may also be responsible for bleeding. Prothrombin time and activated partial thromboplastin time measurements as well as determination of bleeding times are required to assess bleeding diathesis.
Monoclonal light chains can be detected by immunoelectrophoresis in 73% of the urine samples of patients with AL amyloidosis. The λ isotype is twice as frequent as the κ, contrasting with the 1 : 2 ratio of λ to κ observed in myeloma alone. With the use of more sensitive immunochemical techniques, a monoclonal immunoglobulin is found in the serum or the urine in almost 90% of patients. Immunochemical techniques combined with serum free light-chain (FLC) assays detect an abnormal result in 99% of patients.17
AL amyloidosis associated with IgM paraproteinemia characterizes a distinctive subset of patients who have a wider variety of underlying, often lymphoid clonal disorders (including 75% Waldenström macroglobulinemia), usually low-level FLC with a predominance of κ isotype, and higher prevalence of lymph node (31% vs. 3%) and lung (17% vs. 2%) involvement compared with patients with non-IgM monoclonal component.18,19
Diagnosis
AL amyloidosis should be considered in any patient who presents with nephrotic-range proteinuria with or without renal impairment, nondilated cardiomyopathy, peripheral neuropathy, hepatomegaly, or autonomic neuropathy, whether or not a paraprotein can be detected in the serum or urine (Fig. 27-5). Particular vigilance should be maintained in patients with multiple myeloma or monoclonal gammopathy of undetermined significance (MGUS), especially of the λ isotype. Initial investigation should confirm the diagnosis of amyloidosis on tissue biopsy, followed by investigations to establish the type of amyloid present and the extent of organ involvement.
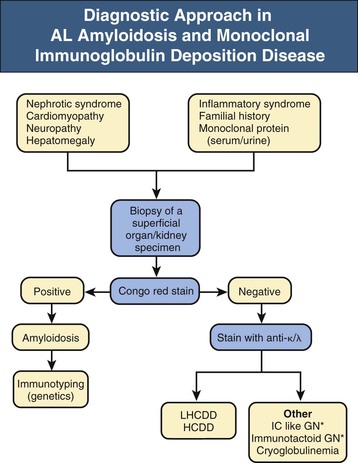
All patients require immunofixation of serum and urine (including serum-free light chains) in an attempt to demonstrate the presence of a monoclonal light chain and quantitation of serum FLC. A bone marrow specimen is necessary because 10% of patients will not have a demonstrable monoclonal light chain by immunofixation, and a clone of plasma cells detected in the bone marrow by immunohistochemistry is strong evidence of AL amyloidosis.
Biopsy of an affected organ is usually diagnostic, but less invasive alternatives should be preferred first. Biopsies of salivary glands or of subcutaneous abdominal fat yield positive results in 80% to 90% of cases. Rectal biopsy is diagnostic in more than 80%, provided the biopsy specimen contains submucosal vessels in which early deposits are located. Bone marrow biopsy specimens should be stained with Congo red for the presence of amyloid, and involvement of the bone marrow (observed in about 50% of patients) is strongly suggestive of the AL type. Evaluation of adequate specimens in experienced laboratories is necessary to maintain high diagnostic sensitivity and specificity.
It is not always easy to be certain that amyloidosis is of the AL type because immunohistochemical staining for immunoglobulin light chains may not be diagnostic, and the presence of a monoclonal component is strong but not conclusive evidence of the AL type. Caution is required when patients have an intact monoclonal immunoglobulin in the serum without evidence of FLC in the serum and urine. In those cases, hereditary forms of amyloidosis should be considered because they may produce clinical syndromes indistinguishable from AL and may coexist with MGUS.9 In cases of doubt, DNA analysis and amyloid fibril sequencing by mass spectrometry are necessary.
Treatment and Outcome
AL amyloidosis is among the most severe complications of plasma cell proliferative disorders. Cardiac involvement responsible for congestive heart failure (CHF) and arrhythmias accounts for at least 40% of deaths. Therapy is aimed at annihilation of the plasma cell clone: “partial response” is now defined by a 50% or greater reduction in the difference between involved FLC (secreted by the plasma cell clone) and uninvolved FLC (the other isotype; dFLC), “very good partial response” by dFLC less than 40 mg/L, and “complete response” by the absence of detectable monoclonal immunoglobulin with normal serum FLC and κ/λ ratio.20 In the responders, gradual regression of AL amyloid deposits is possible. Clinical improvement does not parallel regression of amyloid load.21 Scintigraphy after injection of [123I]-SAP component may be helpful for monitoring the extent of systemic amyloidosis, but this technique is available in a few centers only.
Only 15 years ago, overall survival in patients with AL amyloidosis was poor under a combination of melphalan plus prednisone (MP), compared with no treatment or colchicine therapy alone.22,23 The prognosis of the disease has been transformed with the development of new strategies derived from myeloma treatment. High-dose melphalan followed by autologous stem cell transplantation (HDM/SCT) was introduced,24 but treatment-related mortality (TRM) was consistently higher than in similarly treated patients with multiple myeloma: TRM was 11% for all patients over 15 years, and 5% in the last 5 years, but with improved patient selection and experienced management.25 Despite its efficacy, HDM/SCT in AL amyloidosis generally remains restricted to those less than 65 years old, with up to two organs involved and without cardiac amyloidosis. Its place as first-line therapy in systemic AL amyloidosis is questionable.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


