Primary glomerular disease
Systemic disease
Metabolic disease
De novo disease
Focal segmental glomerulosclerosis
Systemic lupus erythematosus
Diabetic nephropathy
Membranous nephropathy
IgA nephropathy
Henoch–Schonlein purpura
Oxalosis
Anti-GBM disease
MPGN type I
Atypical HUS
Fabry’s disease
HUS
Dense deposit disease
Amyloidosis/LCDD
Diabetic nephropathy
Membranous nephropathy
Anti-GBM disease
Fibrillary GN
ANCA vasculitis
Scleroderma
Single-center and multicenter series have reported recurrent GN rates ranging from 1.8 to 20 %, depending on the criteria used to diagnose recurrence, and the duration of post-transplant follow-up. In the Renal Allograft Disease Registry (RADR), a consortium of six American transplant centers, the rate of recurrent and de novo GN was 3.4 % at a mean follow-up of 65 months [1]. Briganti et al., in analyzing registry data from Australia and New Zealand, found that 8.4 % of allografts were lost due to recurrent GN at 10 years post-transplant [2]. The analysis did not include patients with recurrent disease who did not lose their grafts, but the fact that the incidence of graft loss from recurrent GN continued to increase over time suggests that the overall incidence of recurrent GN in this population was higher than 8.4 %. The risk for recurrence (either clinical or histological) for various diseases is shown in Table 15.2. There have been no registry reports on de novo disease, and in the RADR recurrent and de novo diseases were analyzed together, making it difficult to determine the overall prevalence of de novo disease.
Table 15.2
Risk of disease recurrence
Disease | Risk in percentage |
|---|---|
FSGS | 30–50 |
IgA nephropathy | 30–50 |
MPGN type I | 20–50 |
Membranous nephropathy | 10–40 |
Dense deposit disease | 80–100 |
Atypical HUS | 50 |
ANCA-associated vasculitis | 10–20 |
SLE | 10–30 |
HSP | 20–80 |
Diabetes mellitus | 40 |
Oxalosis | 80–100 |
Fabry’s disease | <5 |
Clinical Presentation
Recurrent and de novo glomerular diseases have similar clinical findings as native kidney disease, with a combination of proteinuria, microscopic or gross hematuria, and/or graft dysfunction. The time to histologic or clinical recurrence of glomerular disease can range from immediately postoperative (focal segmental glomerulosclerosis (FSGS)) to more than a decade after transplant (IgA nephropathy (IgAN) or diabetic nephropathy). True de novo disease, as opposed to recurrence of native kidney disease, tends to occur later after transplant, although cases of de novo anti-glomerular basement membrane (GBM) disease in patients with Alport’s disease have been described in the first month after transplant. The development of drug-induced de novo thrombotic microangiopathy (TMA), due to calcineurin-inhibitors or mTOR inhibitors, usually occurs within the first few months post-transplant.
Impact on Graft Outcomes
Despite advances in understanding the pathogenesis of many renal diseases, treatment options for many recurrent diseases remain limited and disease recurrence frequently leads to allograft dysfunction and ultimately failure. In RADR, the 5-year graft survival in patients with recurrent disease was 39.8 % compared with 67.6 % in patients without recurrent disease. The relative risk for graft failure was 1.9, and the relative risk for graft failure for individual diseases was significantly higher for membranoproliferative GN (MPGN) (2.37), FSGS (2.25), and hemolytic-uremic syndrome (HUS) (5.36) [1]. Analysis of registry data from Australia found an 8.4 % rate of graft failure from recurrent disease over 10 years. In that analysis, the relative risk of graft failure was significantly higher for FSGS (2.03) and MPGN type I (2.91) [2]. For certain diseases, recurrence may be more common with live donors (especially living-related donors) as compared with deceased donors. Indeed, some authors have found that recurrent disease is the most common cause of graft failure in recipients of HLA-identical living-related transplants [3]. However, despite the higher risk of graft failure from recurrent disease, overall graft survival for all primary diseases tends to be higher with living donors as compared with deceased donors. Figure 15.1a shows the relative risk of allograft failure from recurrent disease, as compared with other common causes of graft failure. Figure 15.2 shows the relative risk of allograft failure from recurrent glomerulonephritis for different subtypes of glomerular disease.
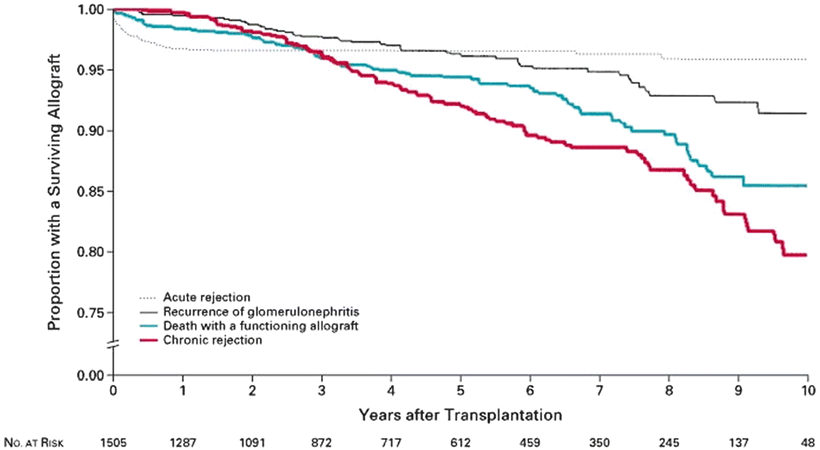
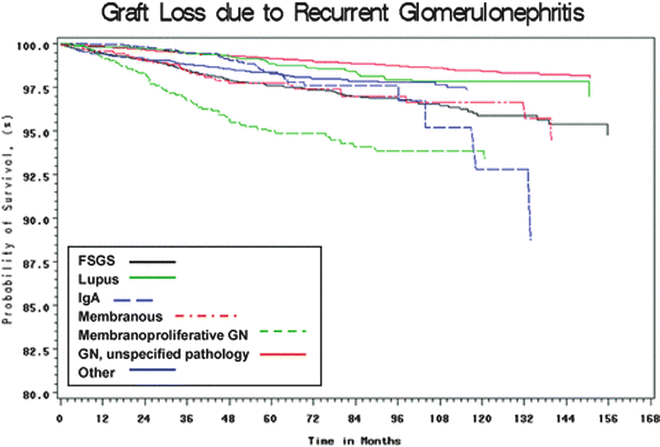
Fig. 15.1
Allograft failure due to recurrent disease, as compared with allograft failure due to other causes. Briganti EM et al. N Engl J Med 2002;347:103-109
Fig. 15.2
Allograft failure from recurrent disease, by subtype of glomerular disease. American Journal of Transplantation. Volume 9, Issue 4, pages 804-811, 6 Mar 2009 DOI: 10.1111/j.1600-6143.2009.02554.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1600-6143.2009.02554.x/full#f1
There are fewer data on the impact of de novo disease on graft survival. Early reports of de novo TMA likely included many patients with antibody-mediated rejection, making it difficult to determine the relative contribution of each to graft failure. De novo TMA, when caused by medications, has an excellent prognosis when the drug is able to be withdrawn completely [4]. Graft loss is rare in patients with de novo TMA localized to the kidney, although it may occur in some patients with systemic manifestations of TMA [5]. De novo diabetic nephropathy progresses more rapidly than native kidney disease and has a poorer prognosis [6]. De novo membranous nephropathy, which may represent a form of chronic rejection (discussed below), has a poor prognosis, with up to 40 % progressing to graft failure [7].
Impact of Immunosuppression on Disease Recurrence
While improvements in induction and maintenance immunosuppression have greatly improved renal allograft outcomes, these advances do not appear to have translated into any decrease in recurrent disease. Analysis of USRDS data from 1990 to 2003 found a 10-year incidence of allograft failure from recurrent GN of 2.6 %. The use of prednisone, cyclosporine, tacrolimus, sirolimus, azathioprine, or mycophenolate was not associated with any difference in graft failure from recurrent disease. Additionally, there was no difference in the risk of graft failure between cyclosporine and tacrolimus or between azathioprine and mycophenolate [8]. Although the choice of immunosuppression medication did not impact the risk of graft failure from recurrent disease, the risk of graft failure from recurrent disease was lower in transplants performed in more recent years.
Although registry data have not shown an increased risk of graft loss from recurrent disease in patients not maintained on prednisone, Kukla et al. reported higher rates of recurrent glomerular disease in patients on a steroid-free regimen: 1-, 5-, and 7-year recurrence rates were 6.7, 13.7, and 19.2%, compared with 2.4, 3.8, and 5.3 % in historic controls maintained on prednisone. Rates of histologic recurrence were significantly higher at all time points for FSGS, membranous, and IgAN, and were numerically, though not statistically, higher at all time points for MPGN. Despite the higher rate of disease recurrence, there was no difference in allograft survival at 7 years [9].
Pascual et al. reported on the effect of induction therapy on the risk of recurrent disease in 443 recipients with ESRD secondary to biopsy-proven glomerular disease. Clinical recurrence of glomerular disease was numerically higher with use of alemtuzumab (8.7 %) or basiliximab (10.1 %) compared with use of thymoglobulin (1.5 %). Mean follow-up was significantly shorter with alemtuzumab, raising concern that larger differences in recurrence rates could develop over time [10].
Pathogenesis and Treatment
The pathogenesis and treatment differ for each recurrent disease. Table 15.3 summarizes current understanding of the pathogenesis of the individual diseases discussed below, as well as potential treatment options based on our current understanding of each disease.
Table 15.3
Possible pathogenetic factors and potential therapeutic interventions used for various recurrent and de novo diseases
Disease | Pathogenesis | Potential therapies |
|---|---|---|
FSGS | Circulating serum factor | Plasmapheresis, rituximab, galactose |
Membranous nephropathy | Anti-PLA2R antibodies | Rituximab |
IgA nephropathy | Aberrantly glycosylated IgA1 | |
MPGN type I | Circulating immune complexes, dysregulation of alternative complement pathway | Eculizumab |
Dense deposit disease | Dysregulation of alternative complement pathway | Eculizumab, rituximab |
Atypical HUS | Dysregulation of alternative complement pathway | Eculizumab |
Lupus | Autoantibodies | |
Anti-GBM disease | Antibody to alpha5 chain of type IV collagen | Plasmapheresis, cyclophosphamide |
ANCA vasculitis | P-ANCA, C-ANCA | Plasmapheresis, cyclophosphamide, rituximab |
Diabetes mellitus | Hyperglycemia | Pancreas transplantation, with or after kidney transplantation |
Primary Glomerular Diseases
Focal Segmental Glomerulosclerosis
FSGS recurs in 20–50 % of patient after transplant. The wide range of reported recurrence rates is likely explained by the fact that some patients had secondary forms of FSGS in the native kidney, which would not be expected to recur. Recurrent FSGS is characterized by the early onset of heavy proteinuria in association with graft dysfunction. Disease recurrence typically occurs within the first year post-transplant. FSGS may recur within the first 24 h of transplant, and foot process effacement may be found on intraoperative biopsies [11]. Biopsies taken early in the course of disease recurrence may appear normal by light microscopy, with histologic evidence of the disease being limited to extensive foot process effacement by EM. However, as the disease progresses, classic lesions of FSGS will become apparent on light microscopy.
The finding that FSGS can recur immediately post-transplant suggested the presence of a circulating factor causing the initial podocyte injury [12]. Savin et al. reported finding a circulating permeability factor in the serum of some patients with recurrent FSGS that increased glomerular permeability to albumin [13]. Recently, Wei et al. found that levels of serum-soluble urokinase receptor (suPAR) are elevated in many patients with primary FSGS and that higher levels pre-transplant increased the risk of post-transplant recurrence. Animal models confirmed the ability of circulating suPAR to activate podocyte β3 integrin, leading to foot process effacement, proteinuria, and FSGS [14].
While these findings suggest that suPAR may be the permeability factor responsible for some cases of recurrent FSGS, recurrent disease can also develop in patients whose FSGS is due to an underlying genetic defect. Congenital nephrotic syndrome of the Finnish type, due to mutations in nephrin, recurs in 25 % of patients. Disease recurrence appears to be limited to patients with the Fin-major/Fin-major genotype, who have complete absence of nephrin in their native kidneys and develop circulating anti-nephrin antibodies directed against the transplant [15]. Jungraithmayr et al. screened 53 patients for mutations in NPHS2. Recurrence rates were 0 % in patients with homozygous, heterozygous, or compound heterozygous mutations in NPHS2 and 45 % in patients without any NPHS2 mutations; graft failure was also significantly higher in patients without NPHS2 mutations [16]. While recent research has identified mutations in the TRPC6, CD2AP, ACTN4, and IFN2 genes leading to FSGS, there are currently no data on whether mutations in these genes affect the risk of disease recurrence after transplant.
The clinical risk factors for recurrent FSGS include younger age, rapid progression of original disease, Caucasian race, and a history of prior graft loss due to recurrent disease. Early reports suggested that graft loss from recurrent FSGS may be higher with living-related donors, especially HLA-identical donors. However, a large registry analysis found that, after correction for other risk factors, a living donor had no association with graft loss from recurrent disease [17]. Further analysis revealed that long-term graft survival was significantly better with 0-mismatch live donors as compared with nonidentical live donors or any type of deceased donor. While long-term graft survival is excellent, at 10 years 12.7 % of grafts may be lost due to recurrent disease, and the risk of allograft failure from recurrent disease is significantly greater with FSGS compared with most other forms of primary glomerular disease [18].
The most commonly used treatment for recurrent FSGS is plasmapheresis, which leads to partial or complete remission in 60–70 % of patients. Plasmapheresis is usually administered three times a week until the patient is in clinical remission, followed by a gradual reduction in the frequency of treatments. While many patients are able to discontinue plasmapheresis without triggering a relapse, some patients require chronic plasmapheresis in order to maintain a clinical remission. The efficacy of plasmapheresis is in theory due to decreasing the concentration of the circulating factor causing podocyte injury. Wei et al. reported that two patients with recurrent FSGS had remissions with plasmapheresis associated with a reduction in suPAR to near physiologic levels, while two patients in whom suPAR remained elevated despite plasmapheresis did not experience remission [14]. While these findings are intriguing, it remains to be determined whether monitoring suPAR levels will be useful in guiding clinical management of recurrent FSGS.
Other treatment options for recurrent FSGS include high-dose steroids and intravenous cyclosporine for 14 days [19]. Rituximab has been tried as either initial or salvage therapy for patients with recurrent FSGS, either at the time of initial recurrence or in the setting of a relapse. In a review of the published literature to-date, complete remission of proteinuria was seen in 43.5 % of patients, with partial remission seen in an additional 20.5 %. However, plasmapheresis was given concurrently in 38/39 patients. On multivariate analysis, younger age and normal serum albumin at the time of recurrence were the only two factors predictive of a response to rituximab [20]. There is a single case report of oral galactose being used successfully to treat recurrent FSGS, possibly by binding to the circulating permeability factor [21].
It is uncertain whether pre-transplant plasmapheresis has any beneficial effect on the likelihood of recurrence. Gohh et al. reported that 7/10 patients considered at high risk for recurrence, including 3/6 patients who had lost a prior graft to recurrent disease, were free of recurrent FSGS and had excellent graft function after receiving eight perioperative plasmapheresis treatments [22]. By contrast, Hickson et al. found that FSGS recurred in 6/7 high-risk patients treated with preemptive plasmapheresis therapy [23]. As these studies did not report suPAR levels, it remains unclear whether pre-transplant plasmapheresis to reduce suPAR levels will impact the risk or timing of recurrent disease.
Immunoglobulin A Nephropathy
IgAN is the most frequent primary glomerulonephritis. IgAN is now understood to be a systemic disease in which patients develop elevated serum levels of galactose-deficient IgA1. Antibodies directed against galactose-deficient IgA1 then form, leading to the development of pathogenic immune complexes, which deposit in the mesangium, and induce cellular activation and glomerular injury [24].
Recurrence rates of IgAN vary considerably depending on the time from transplantation to allograft biopsy, whether recurrence is defined clinically or histologically, and whether histologic recurrence requires evidence of IgAN by light microscopy or requires only evidence of IgA deposits by IF or EM. Ortiz et al. examined 65 protocol biopsies performed a median of 6.9 months after transplant. Defining IgAN as the presence of 1+ mesangial deposits for IgA by IF, they found recurrent IgAN in 32.3 % of patients, with 52 % lacking clinical findings, and 65 % having normal histology by light microscopy [25]. Coppo et al., when including microscopic hematuria plus proteinuria >0.5 g/day in the diagnosis of recurrent disease, found a 50 % recurrence rate by IF in protocol biopsies performed a mean of 2.9 years after transplant [26]. The recurrence rate in biopsies performed for clinical indications ranges from 13 to 50 %, although in some cases recurrent disease was not the predominant explanation for graft dysfunction.
The prevalence of clinical recurrence increases over time, and may be diagnosed more than 10 years after transplant. Recurrence occurs more frequently in younger patients and patients with a more rapid progression of their native kidney disease, including those with underlying crescentic GN [27]. Biopsy-proven recurrent disease is more common in patients who receive a zero HLA-mismatch live donor transplant [28]. The presence of latent IgA deposits in the allograft at the time of transplant (i.e., donor transmitted) may be associated with a higher risk of recurrent disease [29]. Only one study has examined the relationship of aberrantly glycosylated IgA1 to recurrent disease. In a study of 61 patients with histologic recurrence of IgAN, there was no significant increase in the risk of recurrent disease associated with higher levels of aberrantly glycosylated IgA1. However, certain genetic polymorphisms of IL-10 and TNF-α were associated with protection from early recurrence of IgAN [26]. The risk of recurrent disease is higher in patients who lost a prior allograft due to recurrent IgAN. Other pre-transplant characteristics, including gender, ethnicity, and source of allograft, are not consistently predictive of recurrence.
Angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers are often prescribed in an effort to reduce blood pressure and proteinuria in recurrent IgAN. In one Japanese study of recurrent IgAN, 16 patients underwent tonsillectomy. Proteinuria decreased in all 16 patients, compared with no change in proteinuria in 12 patients who did not undergo tonsillectomy [30]. If disease recurrence is associated with a rapid course and the presence of crescents on biopsy, high-dose steroids and cyclophosphamide have been tried, similar to their use in native kidney disease. However, there is no data to confirm their efficacy in recurrent disease.
Due to the categorization of patients with mesangial IgA deposits and no clinical disease as having recurrent disease, overall, recurrence of IgAN has not been shown to increase the risk of allograft failure in the first decade after transplant. Indeed, 5-year graft survival may be better in patients with IgAN compared with other primary diseases. However, with longer follow-up graft survival becomes comparable to and may be worse than other diseases. Ponticelli et al. found slightly lower 10-year graft survival with IgAN when compared with recipients with ESRD from all other causes (75 % vs. 82 %) [27]. Choy et al. reported 17-year graft survival of 61 % compared with 70 % in non-IgAN patients, with the graft survival curves crossing at 12 years [31]. These data suggest that the detrimental impact of recurrent IgAN on graft survival may become more apparent with longer-term follow-up.
Berthoux et al. found a significantly lower rate of clinically recurrent IgAN at 5 years with thymoglobulin (9 %) compared with either IL2-receptor antagonists (41 %) or no induction agent (14 %) [32]. By contrast, Ortiz et al., using protocol biopsies, found histologic recurrence in 28.6 % of patients induced with thymoglobulin at a mean of 6.9 months after transplant [25]. Kukla et al. found that histologic recurrence of IgAN was significantly more common in patients who underwent early steroid withdrawal, with recurrent disease seen in 8.8 % of patients at 5 years compared with 3.3 % of patients maintained on steroids. However, there was no impact on graft survival [9]. By contrast, registry data have showed an increased risk of allograft failure in patients who underwent steroid withdrawal at any time after transplant. However, the absolute risk of graft failure from recurrent disease was low [33]. Taken together, the current data suggest that choice of induction or maintenance immunosuppressive agents plays at most a small role in the risk of graft failure from recurrent IgAN.
Membranous Nephropathy
Membranous nephropathy is characterized histologically by subepithelial immune-deposits in the GBM. Beck et al. have identified circulating autoantibodies directed against the M-type phospholipase A2 receptor in 70 % of patients with idiopathic native kidney membranous nephropathy [34]. Levels of anti-PLA2R antibodies correlate with disease activity, and several recent findings suggest that these autoantibodies are pathogenic [35, 36].
Following transplantation, the histologic recurrence rate of membranous nephropathy may be as high as 42 % at a mean of 13 months post-transplant when patients are evaluated by protocol biopsy. Disease recurrence was identified as early as 2 months post-transplant, and six of eight recurrences occurred in the first year. However, disease recurrence was also identified more than 5 years after transplant [37]. Moroni et al. found a recurrence rate of 34 % in patients who underwent biopsy for proteinuria or graft dysfunction at mean of 26.2 months after transplant [38]. While the rate of clinical recurrence is less than the rate of histologic recurrence, this study suggests that the reported rate of recurrent membranous will continue to rise with longer periods of follow-up.
No identifiable risk factors have consistently been associated with an increased risk of recurrent membranous. Some authors have found an association of recurrent membranous with anti-PLA2R antibodies. Debiec et al. found that five of ten cases of recurrent membranous were associated with anti-PLA2R antibodies. However, they also found that three patients with anti-PLA2R antibodies did not recur. In patients with antibodies present prior to transplant, the titer did not predict time to recurrence [39]. Kukla et al. found a higher rate of clinical recurrence in patients who underwent rapid steroid withdrawal (31 % vs. 10 % at 7 years), although there was no impact on graft survival [9].
Briganti et al. found that overall 12.5 % of patients with membranous nephropathy experienced allograft failure due to recurrent disease over 10 years of follow-up [2]. Moroni et al. found that 50 % of patients who experienced recurrent disease lost their grafts due to disease recurrence [38]. Patients with graft failure from recurrent disease had earlier recurrences and higher levels of proteinuria at the time of diagnosis. However, membranous nephropathy as a cause of ESRD is not associated with an increased risk of graft failure compared with other primary glomerular diseases, and 10-year graft survival is approximately 60% for patients with membranous nephropathy. There are no prospective studies of treatment of recurrent membranous. Several case reports have suggested that rituximab may be beneficial in some patients with recurrent membranous [40, 41]. Other authors have reported success with the use of angiotensin-converting enzyme inhibitors, high-dose steroids, and cyclophosphamide.
Recurrent membranous nephropathy must be distinguished from de novo disease. It is difficult to determine precisely the frequency of de novo membranous, as some cases undoubtedly represent recurrent disease in patients who did not undergo native kidney biopsy. Case series have estimated that de novo membranous occurs in 0.7–9.3 % of recipients [7]. Honda et al. found that de novo membranous occurs late after transplant, at a mean of 102 months, and presents with varying degrees of proteinuria and graft dysfunction. De novo membranous most likely represents a form of alloimmune response, as histologic findings of acute or chronic antibody-mediated rejection were seen in the majority of patients with de novo membranous, and 30 % had evidence of donor-specific antibodies. The outcome of de novo membranous, especially in those who also had donor-specific antibodies, is generally poor [42].
Membranoproliferative Glomerulonephritis
MPGN is a histologically defined disease that is diagnosed based on a characteristic pattern of injury seen on biopsy. Traditionally, MPGN was classified as type I, type II (also known as dense deposit disease (DDD), discussed below), or type III based on EM findings. Recent studies have pointed to the role of the alternative complement pathway in MPGN. Given these findings, a more practical clinical approach may be to view MPGN as either immune-complex-mediated or complement-mediated [43]. While this classification system may better reflect the underlying pathophysiology of MPGN, there are no data on the risk of recurrent disease based on this schema.
The prevalence and course of recurrent MPGN type I were described in several single-center analyses. Moroni et al. reported a recurrence rate of 23.5 %, occurring at a mean of 40 months (range 3.5–105) after transplant [44]. Using protocol biopsies, Lorenz et al. found a recurrence rate of 41.4 %, with only half of the recurrences having clinical findings at the time of diagnosis. All recurrences were diagnosed within the first 14 months after transplant, with the earliest recurrence documented at 1 week post-transplant [45]. Little et al. found a recurrence rate of 49 %, occurring at a median of 4.7 years post-transplant [46]. While MPGN type I may recur early after renal transplant, these data suggest that histologic recurrence may precede clinical recurrence by several years. Reported risk factors for disease recurrence include younger age at initial diagnosis, use of living-related donors, and the degree of mesangial proliferation and crescent formation on native kidney biopsy [44, 46]. In patients who lost a prior allograft to recurrent MPGN type I, the risk of disease recurrence in a second allograft is over 80 %, and nearly all these grafts will fail due to recurrent disease. Developing recurrent MPGN portends a poor prognosis, with graft failure rates ranging from 50 to 67 % [44, 46]. Overall, approximately 15 % of recipients with MPGN will lose their graft to recurrent disease in the first 10 years after transplant, with two-thirds of these failures occurring in the first 2 years after transplant. In registry studies, the hazard ratio for graft loss was 2.91 for patients with MPGN type I compared with other forms of primary GN [2].
Related posts:
 Live Donor Transplantation
Psychosocial Issues in Renal Transplantation
Drug Interactions in Solid Organ Transplant Recipients
Live Donor Transplantation
Psychosocial Issues in Renal Transplantation
Drug Interactions in Solid Organ Transplant Recipients
 Prevention and Management of Infectious Complications in Kidney Transplant Recipients
Prevention and Management of Infectious Complications in Kidney Transplant Recipients
 Urologic Issues in the Renal Transplant Patient
Urologic Issues in the Renal Transplant Patient
 Immunological Assessment of the Transplant Patient
Immunological Assessment of the Transplant Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


