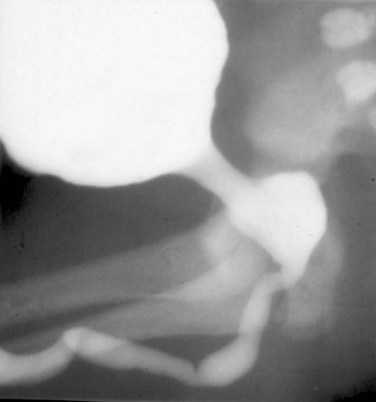
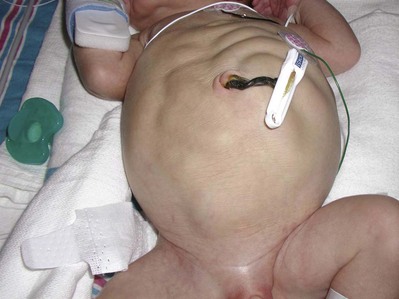
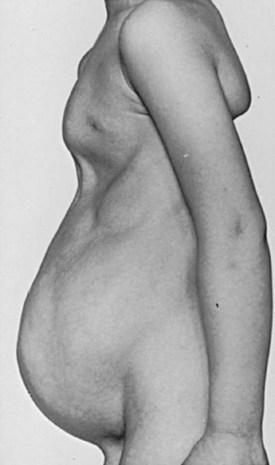
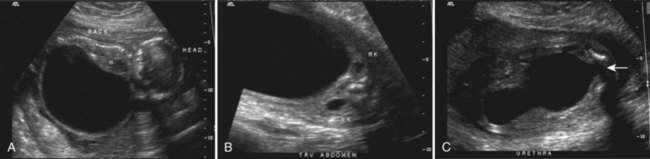
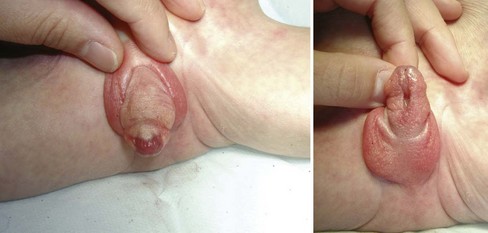
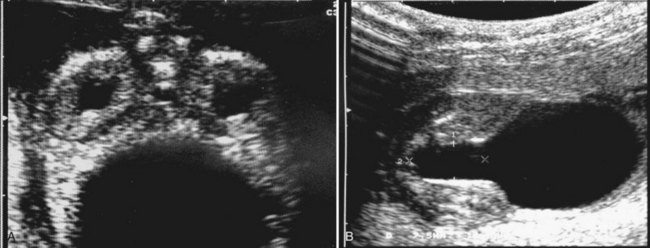
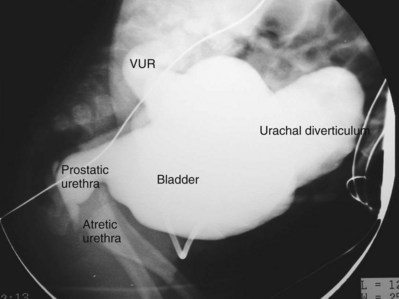
Anthony A. Caldamone, MD, MMS, FAAP, FACS, John R. Woodard, MD The characteristic abdominal wall was first described by Frolich in 1839, and the full triad of anomalies by Parker in 1895. Osler’s vivid description of the abdominal wall of an infant with the characteristic findings led to the term prune-belly syndrome (Osler, 1901). Other names that have been applied to this syndrome include triad syndrome, Eagle-Barrett syndrome, and abdominal musculation syndrome (Eagle and Barrett, 1950; Greskovich and Nyberg, 1988). The incidence of PBS has been reported to be 1 in 29,000 to 1 in 40,000 live births, similar to that of bladder exstrophy (Williams and Burkholder, 1967), with 95% occurring in males (Wheatley et al, 1996). Females with PBS have the abdominal wall deficiency and urinary tract dysmorphism without any gonadal anomaly (Rabinowitz and Schillinger, 1977; Reinberg et al, 1991b). A higher incidence is noted in twins, blacks, and children born to younger mothers. The incidence appears to be declining, possibly because of prenatal diagnosis and a decision to terminate the pregnancy. The high male-to-female ratio, occasional occurrence in male siblings and cousins, and increased occurrence in twins suggest a genetic basis for PBS. Yet most cases are sporadic and have a normal karyotype. One in 23 children with PBS is the product of a twin pregnancy (Ives, 1974). However, the majority of reported twins have been discordant for PBS, evidence against a genetic etiology. It has been suggested that the etiology in twins may be due to an uneven distribution of mesenchymal tissue at a critical time of primitive streak development during the third week of embryogenesis (Coplen et al, 1996). There is a reported association with Turner syndrome, monosomy 16, trisomy 13, and trisomy 18 (Amacker et al, 1986; Hoagland and Hutchins, 1987). A variety of inheritance patterns have been proposed including X-linked recessive (Frydman et al, 1993), a two-step autosomal dominant mutation (Riccardi and Grum, 1977), and polygenetic transmission (Garlinger and Ott, 1974; Lockhart et al, 1979; Adeyokunnu and Familusi, 1982). A report by Ramasamy and colleagues (2005) suggested a sex-influenced autosomal recessive mode of inheritance in familial PBS. The consensus, however, remains that an associated chromosomal abnormality is the exception rather than the rule because most have a normal karyotype. Other reports have noted an association between PBS and Beckwith-Wiedemann syndrome (Silengo et al, 2002; Sinico et al, 2004). Several theories about the embryogenesis of PBS have been proposed. However, because there is no experimental model that can be used to test these theories, the exact mechanism remains elusive. The four chief theories are (1) early in utero posterior urethral obstruction resulting in severe dilation of urinary tract and possible fetal ascites and oligohydramnios (Strumme, 1903; Pagon et al, 1979; Wheatley et al, 1996; Beasley et al, 1988); (2) primary defect in the lateral plate mesoderm, which is the precursor of the ureters, bladder, prostate, urethra, and gubernaculum (Ives, 1974; Gonzalez et al, 1990); (3) an intrinsic defect of the urinary tract leading to ureteral dilation and fetal ascites (Symonds and Driscoll, 1974; Monie and Monie, 1979; Smythe, 1981; Nakayama et al, 1984; Cazorla et al, 1997); and (4) a yolk sac defect (Stephens, 1983; Stephens and Gupta, 1994). None of these theories have universal acceptance, and there is some overlap between them. A spectrum of renal abnormalities extends from normal renal parenchyma to dysplasia (Fig. 123–1). The more severely dysplastic kidneys are generally associated with bladder outlet obstruction in which there has not been decompression through a patent urachus (Potter, 1972). Dysplasia is present in 50% of cases; however, it may vary in degree and laterally (Rogers and Ostrow, 1973; Stephens, 1983). Renal dysplasia in PBS of the Potter type II and IV varieties is seen. The Potter type II variety with few nephrons and parenchymal disorganization is more indicative of a renal mesenchymal defect, whereas the Potter type IV with cortical and tubular cysts is associated with outlet obstruction (Wigger and Blanc, 1977). The renal collecting system is characteristically dilated, often to a severe degree. The degree of dilation, however, does not correlate with the degree of renal dysplasia. Calyceal morphology may be well preserved, even in the presence of massively dilated ureters and renal pelves (Berdon et al, 1977). Ureteropelvic junction obstruction can occur on a primary or secondary basis; however, nonobstructive hydronephrosis is the rule (Woodard and Parrott, 1978b). It is renal infection rather than obstruction that poses the greatest risk to renal function. The ureters are typically dilated, tortuous, and redundant (Fig. 123–2). The proximal (upper) portions of the ureters are usually less abnormal than the distal segments, although massive dilation and stenosis can occur at all levels. Histologic sectioning demonstrates a lack of smooth muscle cells and an increase in fibrous connective tissue. Generally there are more normal-appearing smooth muscle cells in the proximal segments (Palmer and Tesluk, 1974; Stephens, 1983). This fact is critical when ureteral reconstruction is undertaken. The ratio of collagen to smooth muscle cells in prune-belly ureters has been noted to be elevated, especially in refluxing ureters (Gearhart et al, 1995). A decreased number of thick and thin myofibrils noted on ultrastructural examination is thought to contribute to the poor peristalsis (Berdon et al, 1977; Stephens, 1983). Vesicoureteral reflux is present in 75% of children with PBS (Berdon et al, 1977; Fallat et al, 1989) (Fig. 123–3). Obstruction is not common but has been reported at both the ureteropelvic and ureterovesical junctions (Wigger and Blanc, 1977; Moerman et al, 1982; Manivel et al, 1989). These large ureters may have ineffective peristalsis because of poor ureteral wall coaptation. The ureteral conduction wave reaches a reduced smooth muscle cell population of poor contractile potential related to reduced myofibrils, often separated by patches of collagen with a resulting bolus of urine reaching more dilated ureteral segments as it progresses toward the bladder (Woodard and Smith, 1998). This can be seen fluoroscopically as ineffective peristalsis, resulting in upper tract stasis, which may lead to infection (Nunn and Stephens, 1961; Williams and Burkholder, 1967). Key Points Clinical Features The bladder usually appears massively enlarged with a pseudodiverticulum at the urachus (Fig. 123–4). The urachus is patent at birth in 25% to 30% of children (Lattimer, 1958; Wigger and Blanc, 1977; Stephens and Gupta, 1994). Histologically, the bladder has an increased ratio of collagen to muscle fibers in the absence of obstruction (Workman and Kogan, 1990). The wall is smooth, unlike that seen in obstructed bladders. The pelvic distribution of ganglion cells has been shown to be normal (Nunn and Stephens, 1961; Burke et al, 1969). Smooth muscle hypertrophy is seen, however, in the obstructed prune bladder (Perlmutter, 1976). Stephens demonstrated that the trigone is splayed with the ureteral orifices displaced laterally and superiorly, possibly contributing to the high incidence of reflux (Williams and Burkholder, 1967). On voiding, the bladder neck opens widely into a dilated prostatic urethra (see Fig. 123–4). Urodynamic assessment generally shows normal compliance; however, there is a delayed first sensation to void and large capacity (Snyder et al, 1976). The ability to empty the bladder is variable, with some emptying well and others carrying a significant postvoid residual. This is thought to be due to a relative outlet obstruction and the ability of the bladder to generate sufficient pressure with a detrusor contraction. When the relative outflow resistance prevents effective bladder emptying, the term unbalanced voiding is used (Snyder et al, 1976; Kinahan et al, 1992). Despite these limitations, about 50% of prune-belly patients void spontaneously with normal voiding pressures, normal flow rates, and low postvoid residuals (Nunn and Stephens, 1961; Kinahan et al, 1992). However, as Kinahan and colleagues (1992) demonstrated, deterioration of balanced voiding can occur, resulting in significant postvoid residuals, emphasizing the need for periodic assessment. The dilation of the posterior urethra is due to prostatic hypoplasia, probably related to abnormal mesenchymal-epithelial development (Stephens and Gupta, 1994). Histologically, there are few prostatic cellular elements with a reduction of both epithelial and smooth muscle cells and an increase in connective tissue cells (Moerman et al, 1982; Popek et al, 1991; Stephens and Gupta, 1994). Various obstructive lesions of the distal posterior urethra have been described—urethral atresia, valves, urethral stenosis, urethral membrane, and urethral diverticulum—and are thought to occur in 20% of cases (Hoagland and Hutchins, 1987). Stephens (1983) described an angulation of the urethra during voiding, referred to as type IV valves, which results from lack of prostatic parenchymal tissue. Prostatic hypoplasia, the etiology of which is controversial, is thought to be a factor in the ejaculatory failure of PBS patients (Volmar et al, 2001). The vas deferens and seminal vesicles are often atretic, although either may be dilated or thickened (Stephens and Gupta, 1994). The epididymis may be poorly attached to the testis, as is seen commonly in abdominal undescended testes. There may also be lack of continuity between the efferent ductules and the rete testis. Ejaculation is usually in a retrograde fashion because of an incompetent bladder neck. Although the anterior urethra of the PBS child is usually normal, several anomalies of the anterior urethra have been reported, the most common being urethral atresia and megalourethra (Kroovand et al, 1982; Perrotin et al, 2001). Unless it is associated with a patent urachus, urethral atresia is often lethal (see Fig. 123–3). It has been postulated that urethral atresia or microurethra occurs because the urethra is unused rather than malformed. Spontaneous bladder rupture with fistula formation has also been reported (Reinberg et al, 1993). PBS is associated with two variations of megalourethra (Shrom et al, 1981; Mortensen et al, 1985). The fusiform type is a deficiency of the corpus cavernosum, as well as the spongiosum, and the scaphoid variety is a deficiency of the spongiosum only with preservation of the glans and corpora cavernosa (Fig. 123–5). With the scaphoid variety, the ventral urethra dilates with voiding, whereas with the fusiform variety the entire phallus dilates with voiding. The fusiform variety is thought to result from a mesenchymal deficiency of the urethral folds, whereas the scaphoid variety results from a mesenchymal deficiency of the urethral supportive tissues (Dorairajan, 1963). Megalourethra is more commonly seen in PBS than any other syndrome (Appel et al, 1986). Transient in utero obstruction of the junction between the glanular and penile urethra has been proposed to cause megalourethra. Figure 123–5 Scaphoid megalourethra with hypospadiac orifice of prune-belly syndrome. (Courtesy of C. Peters.) Bilateral intra-abdominal testes lying over the iliac vessels are the most typical findings. Although mechanical forces such as a distended bladder and intra-abdominal pressure have been implicated in maldescent of the testes (Kaplan et al, 1986; Hutson and Beasley, 1988), the fact that some patients with the typical urinary tract and abdominal musculature anomalies (termed pseudoprune patients) may have descended testes raises some doubt about pure mechanical factors. Pak and colleagues (1993) compared the histology of the testes in PBS patients with that of non–prune-belly intra-abdominal testes and that of age-matched control subjects. They found no difference in germ cell counts, Ad spermatogonia, and Leydig cells between PBS testes and non-PBS intra-abdominal testes. However, because germ cell counts in PBS patients younger than 1 year are similar to those of age-matched controls, the implication is that the environmental state of the abdomen is a major factor in their later spermatogenic potential (Nunn and Stephens, 1961; Coplen et al, 1996). This mirrors findings of Nunn and Stephens (1961) of normal germinal epithelium of fetal and newborn PBS testes. Alternatively, Orvis and colleagues (1988) noted decreased numbers of spermatogonia and Leydig cell hyperplasia in fetal PBS testes, implying an intrinsic testicular abnormality. Azoospermia was found in adult PBS patients, and no PBS patient has been reported to have fathered a child (Woodhouse and Snyder, 1985). More recently, Ross and colleagues (1998) have documented paternity in three adults with classic PBS, achieved by sperm retrieval techniques and intracytoplasmic sperm injection. The infertility is thought to be due to a combination of testicular histologic abnormalities, structural defects of the ducts, and prostatic abnormalities (Tayakkanonta, 1963). Three cases of testis tumor have been reported (Woodhouse and Ransley, 1983; Sayre et al, 1986; Massad et al, 1991; Parra et al, 1991). Massad and colleagues (1991) described histologic testicular patterns similar to those in intratubular germ cell neoplasia in three infants. Although the risk of malignancy may be relatively low considering the lack of germinal epithelium (Uehling et al, 1984), it is clear that placement of the testis in the scrotum and long-term follow-up are necessary to reduce the risk of testicular malignancy and enhance detection. Of all children with PBS, 75% have non–urinary tract abnormalities (Geary et al, 1986). After the obvious abdominal wall defect, the most common are cardiac, pulmonary, and orthopedic (Table 123–1). Table 123–1 Extragenitourinary Abnormalities Modified from Woodard JR, Smith EA. Prune-belly syndrome. In: Walsh PC, Retik AB, Vaughan Jr ED, Wein AJ, editors. Campbell’s urology. 7th ed. Philadelphia: WB Saunders; 1998. p. 1991. The appearance of the abdominal wall gives PBS of newborns their most characteristic feature (Fig. 123–6). Although in some cases the musculature of the abdominal wall may be totally absent (Manivel et al, 1989), most commonly there is uneven involvement with the medial and inferior muscular typically most deficient (Mininberg et al, 1973; Randolph, 1977). The appearance at birth is that of wrinkled, redundant skin with an abdomen that bulges in the flanks. One may be able to discern intra-abdominal organs through the thinned abdominal wall. The most severely affected areas may have skin, subcutaneous fat, and a single fibrous layer on the peritoneum (Mininberg et al, 1973; Baird and Sadovnick, 1987). Randolph conducted electromyographic mapping and demonstrated the inferior and medial segments to be most consistently affected (Randolph et al, 1981a). Electron microscopy has demonstrated a nonspecific pattern of myofilament disarray, Z-line disorganization, and mitochondrial proliferation (Afifi et al, 1972; Randolph et al, 1981b; Woodard and Smith, 1998). The fact that normal spinal anterior horn cells have been shown rules out a neuropathic etiology for the muscular deficiency (Nunn and Stephens, 1961). The muscular deficiency, however, is typically inconsistent and patchy. As the child grows older, the abdomen becomes less wrinkled and takes on more of a pot-bellied appearance (Fig. 123–7). Gait is usually not affected, although it may be delayed, and the children tend to roll to their side and use their arms to sit from a supine position. The poor support of the lower chest wall results in flaring of the costal margin (Woodard and Smith, 1998). These children are more vulnerable to respiratory illness because their cough effectiveness is compromised. In spite of these abdominal wall issues, Woodard and Smith (1998) reported good wound healing without a tendency toward infections or incisional hernias. Cardiac anomalies such as patent ductus arteriosus, atrial septal defect, ventricular septal defect, and tetralogy of Fallot occur in 10% of children with PBS (Adebonojo, 1973). Cardiac abnormalities at birth may take precedence over urologic issues. Pulmonary difficulties can be observed at any age in patients with PBS. Pulmonary hypoplasia can result from severe oligohydramnios related to renal dysplasia or severe bladder outlet obstruction and may result in newborn demise. In addition, pneumothorax and pneumomediastinum can be seen with or without pulmonary hypoplasia (Skoog, 1992). Significant pulmonary difficulties have been reported in 55% of PBS survivors (Geary et al, 1986). The lack of ability to generate significant intra-abdominal pressure may contribute to pneumonia and lobar atelectasis (Alford et al, 1978; Ewig et al, 1996). Acute respiratory illnesses or an anesthetic procedure can easily lead to respiratory insufficiency in the PBS patient, who may have underlying chronic bronchitis from repeated respiratory illnesses. Many patients demonstrate significant restrictive lung disease secondary to musculoskeletal abnormalities such as scoliosis, rib cage abnormalities, and compromised abdominal musculature (Coplen et al, 1996). In at least 30% of cases, gastrointestinal anomalies are observed. The majority of the anomalies result from incomplete rotation of the midgut giving way to a wide mesentery, which results in increased bowel mobility with intestinal malrotation, volvulus, atresias, and stenosis (Silverman and Huang, 1950; Wright et al, 1986). Splenic torsion related to abnormal mesenteric fixation has also been reported (Heydenrych and Du Toit, 1978; Teramoto et al, 1981). Omphalocele, gastroschisis, and anorectal abnormalities have been reported (Petersen et al, 1972; Morgan et al, 1978; Wilbert et al, 1978; Short et al, 1985; Walker et al, 1987). With a limited ability to generate intra-abdominal pressure, constipation becomes a lifelong problem and leads to acquired megacolon (Woodard and Smith, 1998). Orthopedic abnormalities, ranging in incidence from 30% to 45%, are second in frequency to those of the genitourinary tract and abdominal wall. Many of these abnormalities result from the compressive effects of oligohydramnios. Some think the musculoskeletal defects result from the abnormal mesenchymal development at 6 weeks of gestation (Loder et al, 1992). Green and colleagues (1993), however, pointed out that because many of the deformities are unilateral, oligohydramnios is the most likely etiology. Dimpling of the lateral aspect of the knees is a common finding in oligohydramnios. Oligohydramnios may also result in talipes equinovarus (26%), hip dysplasia (5%), and congenital scoliosis (4%) (Woodard and Smith, 1998). It has been proposed that a distended bladder that may impinge on the external iliac vessels may compromise the blood supply to the lower extremities and in severely affected cases result in lower extremity hypoplasia, absence, or amputation (Smith, 1913; Green et al, 1993). Prenatal ultrasonography has played a major role in the identification of congenital genitourinary abnormalities. Fetal hydronephrosis can be diagnosed accurately in the second trimester and is present in approximately 1% of all pregnancies. However, the etiology of the hydronephrosis cannot be accurately determined in all cases. Elder (1990) estimated that the accuracy of determining the etiology of fetal hydronephrosis varies from 30% to 85%. In particular, PBS presents prenatally with findings similar to those of other causes of bladder outlet obstruction (Fig. 123–8), as in posterior urethral valves, or megacystis megaureter syndrome (Kramer, 1983). Although an accurate diagnosis of PBS has been made as early as 11 to 14 weeks of gestation (Shimizu et al, 1992; Yamamoto et al, 2001) (Fig. 123–9), the classic findings of hydroureteronephrosis, a distended bladder, and an irregular abdominal circumference are not consistently seen at 30 weeks (Okulski, 1977; Bovicelli et al, 1980; Christopher et al, 1982; Shih et al, 1982). Early fetal ascites has been suggested to correlate with PBS (Scarbrough et al, 1988). It is imperative to remember that the majority of patients with PBS do not have demonstrable urinary obstruction and that the degree of hydroureteronephrosis does not correlate with the postnatal renal function (Gadziala et al, 1982). (Courtesy of E. Ruiz.) Whereas some have recommended in utero intervention for relief of urinary tract dilation and oligohydramnios (Gadziala et al, 1982; Glazer et al, 1982; Nakayama et al, 1984; Scarborough et al, 1988; Estes and Harrison, 1993; Leeners et al, 2000), others have recommended termination of pregnancy (Pescia et al, 1982). It is difficult to justify the advocacy of pregnancy termination in light of our inability to diagnose precisely the etiology of prenatal hydronephrosis and inability to predict postnatal renal function on the basis of the degree of urinary tract dilation, except in rare cases of early and severe oligohydramnios. Prenatal intervention has been applied to PBS with no proven benefit in terms of postnatal renal function (Elder et al, 1987; Sholder et al, 1988; Freedman et al, 1999; Biard et al, 2005; Blaicher et al, 2005). The only circumstances in which prenatal intervention may be justified are the rare situations of urethral atresia with progressive oligohydramnios (Steinhardt et al, 1990; Reinberg et al, 1993; Perez-Brayfield et al, 2001) or cases in which decompression of the urinary tract is necessary to prevent dystocia (Gadziala et al, 1982). The appearance of the abdominal wall immediately suggests the diagnosis of PBS (see Fig. 123–6) whether or not the diagnosis was suspected prenatally. It should be remembered that other associated abnormalities such as cardiac or pulmonary often should take precedence over the urinary tract because in the absence of true bladder outlet obstruction, as seen with urethral atresia, the hydroureteronephrosis is not life threatening. With the number of variable anomalies present in PBS, it is understandable that there is a wide spectrum of clinical presentations. There are three major categories of presentation in the neonatal period as described by Woodard (1985)
Genetics
Embryology
Clinical Features of Prune-Belly Syndrome
Genitourinary Anomalies
Kidneys
Ureters
Bladder
Prostate and Accessory Sex Organs
Anterior Urethra
Testes
Extragenitourinary Abnormalities
Cardiac
Patent ductus arteriosus
Ventricular septal defect
Atrial septal defect
Tetralogy of Fallot
Pulmonary
Lobar atelectasis
Pulmonary hypoplasia
Pneumothorax
Pneumomediastinum
Gastrointestinal
Intestinal malrotation
Intestinal atresias or stenosis
Omphalocele
Gastroschisis
Hirschsprung disease
Imperforate anus
Hepatobiliary anomalies
Orthopedic
Pectus excavatum, pectus carinatum
Scoliosis
Sacral agenesis (partial)
Congenital hip subluxation or dislocation
Genu valgum
Talipes equinovarus
Severe leg maldevelopment
Miscellaneous
Splenic torsion
Adrenal cystic dysplasia
Abdominal Wall Defect
Cardiac Anomalies
Pulmonary
Gastrointestinal Abnormalities
Orthopedic
Presentation
Prenatal Diagnosis and Management
Neonatal Presentation
Spectrum of Disease
Related posts:
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Surgery for Bladder Cancer
Surgery for Bladder Cancer
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Prune-Belly Syndrome
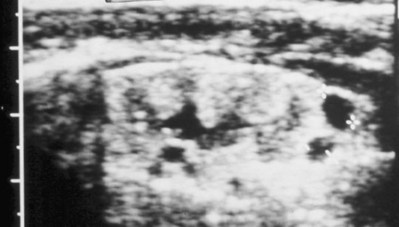
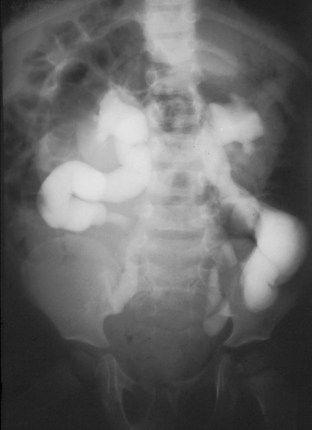
• Hydroureteronephrosis is often to a severe degree; however, the calyceal morphology may be well preserved.