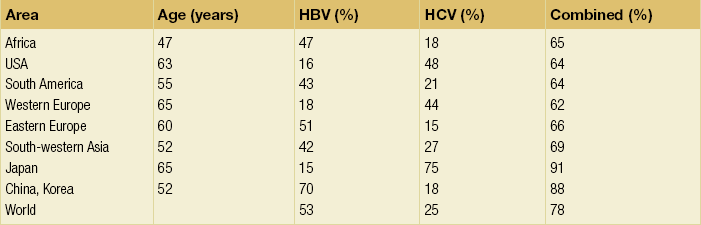
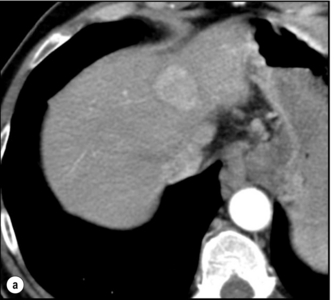
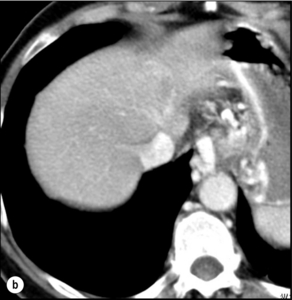
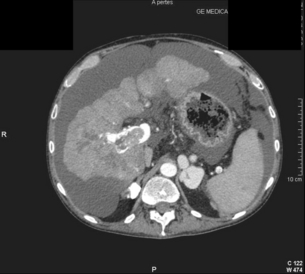
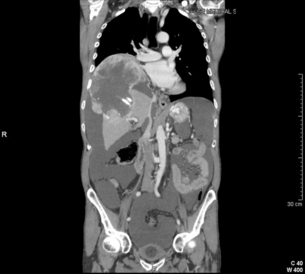
5 HCC accounts for 90% of all primary liver malignancy and its incidence continues to increase. It is the sixth most common neoplasm, accounting for more than 5% of all cancers, and is the third most common cause of cancer-related death. The International Agency for Research on Cancer has estimated in 2008 through its GLOBOCAN series that primary liver cancer caused more than 690 000 deaths worldwide, similar to colon or rectal cancer.1 Control of HCC nodules may be achieved successfully by surgical resection and by percutaneous treatment. The indications for these therapies depend on the morphological features of the tumour and the functional status of the non-tumorous liver. Unfortunately, these treatments are associated with a high incidence of tumour recurrence due to the persistence of the underlying cirrhosis, which represents a preneoplastic condition. Liver transplantation may seem a logical alternative treatment but has its own limitations, including tumour recurrence, the limited availability of grafts, and cost. The most exciting areas of progress are the control of hepatitis B virus (HBV) or hepatitis C virus (HCV), the prevention of carcinogenesis in patients with chronic liver disease, early radiological screening and the development of medical therapies. In the setting of liver surgery, better liver function assessment and understanding of the segmental liver anatomy with more accurate imaging evaluation are the most important factors that have led to a decrease in postoperative mortality. Active follow-up and treatment of recurrence have also contributed to increased 5-year survival rates as high as 70%.2 The world age-adjusted incidence of HCC in men is 14.9 per 100 000, but has geographical variation related to the prevalence of HBV and HCV infections, which are the two main risk factors worldwide and account for more than three-quarters of all cases (Table 5.1). The incidence may be as high as 99 cases per 100 000 in Mongolian men; other high-rate areas include Senegal, Gambia, South Korea, Hong Kong and Japan. By contrast, North and South America, Northern Europe and Oceania are areas with low incidences (less than 5 cases per 100 000); in these areas, HCV is the main risk factor, together with alcohol abuse, non-alcoholic fatty liver disease and obesity. Southern European countries have intermediate rates. The risk of HBV-associated HCC increases with the severity of the underlying hepatitis, age at infection and duration of infection, as well as level of viral replication. An Asian patient with HBV-related cirrhosis has a 17% cumulative risk of developing HCC over a 5-year period. In the West, this cumulative risk is 10%. This may be explained by the earlier acquisition of HBV in Asia through vertical transmission (rather than horizontal transmission in the West through sexual or parenteral routes), longer duration of disease, or additional exposure to environmental factors. Ongoing HBV replication or hepatitis B e-antigen (HBeAg) infection accelerates the progression to cirrhosis and also to HCC. A study conducted in Taiwanese men reported that the risk of HCC increased 10-fold when HBsAg was present and 60-fold when HBeAg was present. Similarly, HBV-DNA levels greater than 104 or 106 copies/mL are associated with a 2.3 and 6.1 hazard risk, respectively, compared to patients with lower levels of replication.3 Additional cofactors that increase the risk of HCC are male gender (three- to sixfold), age > 40 years, concurrent HCV infection (twofold), HDV co-infection (threefold), heavy alcohol consumption (two- to threefold) and, in endemic regions, aflatoxin ingestion. Because anti-HCV vaccination is not available, prevention of HCV infection and minimising the risk of progression of chronic HCV infection to cirrhosis using antiviral treatment is the only means to reduce the incidence of HCV-related HCC. Sustained virological response in HCV-infected patients is associated with a significantly decreased risk of developing HCC.4 The incidence of HCC is expected to rise in HIV-positive persons predominantly because of the higher prevalence of associated well-known risk factors: not only co-infection with HCV and HBV, but also alcohol abuse, non-alcoholic steatohepatitis (NASH) and diabetes. HIV-positive patients who are co-infected with HBV or HCV may have more rapidly progressive liver disease, and when they develop cirrhosis they also have an increased risk of HCC. The Mortavic study indicated that HCC caused 25% of all liver-related deaths among HIV patients.5 Cirrhosis and HCC occur 15–20 years earlier in HIV–HCV co-infected patients than in patients infected by HCV alone. The course of the disease is also considered more aggressive.6 Screening for HCC should, however, be the same as in HIV-negative patients. NAFLD (and the metabolic syndrome) accounts for a substantial portion of what was considered in the past as cryptogenic cirrhosis and carries an inherent risk for the development of HCC. Obesity has definitely been established as a risk factor for the development of HCC, with a 1.5–4 times increased risk.7 An association between NAFLD and HCC was first identified in 2002 by several studies focusing on HCC patients with chronic liver disease in the absence of HBV/HCV infection or alcohol abuse. In this population, there was a much higher prevalence of obesity, diabetes, hypertriglyceridaemia and pathological features of NAFLD. At the same time, evidence was accumulating linking common features of the metabolic syndrome/NASH with HCC. In particular, obesity was noted to increase the mortality from liver cancer far more than for any other cancer.7 Similarly, diabetes was found to increase the risk of HCC with and without acute or chronic liver disease.8 Precise figures on the incidence of HCC in patients with NAFLD are still lacking. It increases with male gender, increasing age, sinusoidal iron deposition and severity of underlying liver disease. In surgical series, overt cirrhosis is present in only one-third of patients while the others have less severe liver damage.9 In addition, there is also evidence that NAFLD may act synergistically with other risk factors, such as chronic HCV or alcoholic consumption, in the development of HCC. Hereditary haemochromatosis (HH) is an autosomal recessive disorder associated with homozygosity for the C282Y mutation in the haemochromatosis gene and characterised by excessive gastrointestinal absorption of iron. HH is a long-known risk factor for HCC, and the risk increases in patients with cirrhosis. Other risk factors include male gender and diabetes. Several additional risk factors such as HBV infection (4.9-fold), age greater than 55 years (13.3-fold) and alcohol abuse (2.3-fold) may act synergistically with iron overload to increase the risk of HCC in patients with cirrhosis caused by hereditary haemochromatosis. In a recent meta-analysis of nine studies including 1102 HCC cases, mainly from European populations, it has been reported that C282Y mutation was associated with increased risk of HCC (fourfold) in patients with alcoholic liver cirrhosis, but not in those with viral liver cirrhosis.10 Interestingly, pathological conditions other than haemochromatosis that are associated with iron overload, such as homozygous β-thalassaemia or the so-called African overload syndrome, are also associated with an increased risk of HCC. Similarly, there is evidence of a link between iron deposits within the liver and HCC in patients with and without cirrhosis. Primary biliary cirrhosis (PBC) has been considered as a low-risk factor for HCC, not only because of its rare incidence but also because it predominantly affects women (with a sex ratio of 9:1). A recent meta-analysis of 12 studies has reported that PBC is significantly associated with an increased risk of HCC (18.8-fold) compared to the general population.11 However, there were several confounding factors in this meta-analysis, such as advanced histological stage of PBC, history of blood transfusion, and smoking or drinking habits that might be associated with an increased probability for HCC development in PBC patients, or may be directly associated with PBC development. In contrast, HCC development in patients with secondary biliary cirrhosis is exceptional if it even exists. Autoimmune hepatitis has a low risk of HCC development. Potential reasons are the female predominance and the delayed development of cirrhosis through corticosteroid therapy. HCV infection needs to be ruled out as it may induce autoantibodies. Recent data reported that cirrhosis at presentation is an important prognostic risk factor for HCC. In a prospective multicentre cohort study evaluating 193 Japanese patients with autoimmune hepatitis, seven (3.6%) developed HCC during an 8-year period, all of whom had underlying cirrhosis.12 Like adenoma in other locations, hepatocellular adenomas (HCAs) have a risk of malignant transformation and hepatocyte dysplasia is the intermediate step between HCAs and HCC. A recent systematic review estimated the risk to be 4.2%.13 This risk and the treatment strategy to prevent it may, however, be refined. HCAs are most classical in women of child-bearing age and are associated with the prolonged use of contraceptives and oestrogen treatments. Discontinuation of oral contraceptives does not completely avoid the risk of malignant transformation. Malignancy within HCAs measuring less than 4 cm in diameter is exceptional. There is also recent evidence that HCAs may develop in men, especially if there is a background of a metabolic syndrome. The risk of malignant transformation in men is 50% (10 times higher than in women) and malignancy can occur in HCAs as small as 1 cm.14 Therefore, whereas resection of HCAs larger than 4 cm is recommended in women, all HCAs irrespective of size should be resected (or ablated) in men. The number of HCAs does not appear to increase the risk of malignant transformation and, in particular, patients with polyadenomatosis are not at increased risk.14–16 Malignant transformation of HCAs has also been linked to the genotype and phenotype of HCAs. It is more prevalent in telangiectatic or atypical HCAs than in steatotic HCAs. Most importantly, the presence of a β-catenin mutation (observed in approximately 10–15% of HCAs) confers a particularly high risk of malignancy.17 Microscopically, HCCs exhibit variable degrees of differentiation that are usually stratified into four different histological grades, known as Edmondson grades 1–4, which correspond to well-differentiated, moderately differentiated, poorly differentiated and undifferentiated types. The degree of differentiation typically decreases as the tumour increases in diameter. Very-well-differentiated HCCs can resemble normal hepatocytes and the trabecular structure may reproduce a near normal lobar architecture so that histological diagnosis by biopsy or following resection may be difficult. A number of immunomarkers have been described to selectively identify the malignant nature of these HCCs, not only in resected specimens but also in liver biopsies: glypican 3 (GPC3), heat shock protein 70 (HSP70) and glutamine synthetase (GS). Positive immunomarker staining for any two markers can detect early and well-differentiated HCC in 50–73% of cases, with 100% specificity when the analysis is performed on resected specimens.18 HCC has a great tendency to spread locally and to invade blood vessels. The rate of portal invasion is higher in the expansive type, in poorly differentiated HCCs and in large tumours. Characteristically, microscopic vascular invasion is seen in 20% of tumours measuring 2 cm in diameter, in 30–60% of cases with nodules measuring 2–5 cm and in up to 60–90% when nodules are more than 5 cm in size. The presence of portal invasion is the most important predictive factor associated with recurrence. The tumour thrombus has its own arterial supply, mainly from the site of the original venous invasion. Once HCC invades the portal vein, tumour thrombi grow rapidly in both directions, and in particular towards the main portal vein. As a consequence, tumour fragments spread throughout the liver as the thrombus crosses segmental branches. Once the tumour thrombus has extended into the main portal vein, there is a high risk of complete thrombosis and increased portal hypertension. This accounts for the frequent presentation with fatal rupture of oesophageal varices, or liver decompensation including ascites (Fig. 5.1), jaundice and encephalopathy. Invasion of hepatic veins is possible, although less frequent. The thrombus eventually extends into the suprahepatic vena cava or the right atrium and is associated with a high risk of lung metastases. Rarely, HCC may invade the biliary tract and give rise to jaundice or haemobilia. Mechanisms of HCC-induced biliary obstruction include: Figure 5.1 CT scan of a patient with a tumour thrombus originating from an HCC located in the right liver. The thrombus extends in to the main portal vein. Ascites is present. • intraductal tumour extension; • obstruction by a fragment of necrotic tumour debris; • haemorrhage of the tumour resulting in haemobilia; • metastatic lymph node compression of major bile ducts in the porta hepatis. Spontaneous rupture occurs in 5–15% of patients and is observed particularly in patients with superficial or protruding tumours. The diagnosis should be suspected in patients with known HCC or cirrhosis presenting with acute epigastric pain, as well as in Asian or African men who develop an acute abdomen (Fig. 5.2). Minor rupture manifests as abdominal pain or haemorrhagic ascites, and hypovolaemic shock is only present in about half of the patients. Portal vein invasion may manifest as upper gastrointestinal bleeding or acute ascites, and invasion of hepatic veins or the inferior vena cava may result in pulmonary embolism or sudden death. Figure 5.2 CT scan of a patient with a ruptured HCC. Note that the rupture is limited at the upper part of the liver. This patient had haemorrhagic ascites. α-Fetoprotein: Serum α-fetoprotein (AFP) is the most widely recognised serum marker of HCC. It is secreted during foetal life but residual levels are very low in adults (0–20 ng/mL). It may increase in patients with an HCC, and serum levels greater than 400 ng/mL can be considered as diagnostic of HCC with 95% confidence. Levels may exceed 10 000 ng/mL in 5–10% of patients with HCC. Very high levels usually correlate with poor differentiation, tumour aggressiveness and vascular invasion. An AFP > 20 ng/mL has a sensitivity of 60% and therefore a surveillance programme using this cut-off value would miss 40% of tumours. If a value of > 200 ng/mL is used, 22% of tumours would be missed. Only 10% of small tumours are associated with raised AFP levels, whereas 30% of patients with chronic active hepatitis without an HCC have a moderately increased AFP. This usually correlates with the degree of histological activity and raised levels of transaminase, and it may therefore fluctuate. Tumours other than HCC can also be associated with increased AFP levels, but these are rare (non-seminal germinal tumours, hepatoid gastric tumours, neuroendocrine tumours). Others serum tumour markers: Alternative serum markers for HCC, such as des-γ-carboxy prothrombin (DCP) or prothrombin induced by vitamin K absence (PIVKA-II; > 40 mAU/mL) and AFP-L3 (> 15%) have not come into common practice except in Japan, where they are covered under the national health insurance. A prospective study of at-risk patients comparing the accuracy of AFP and DCP in the early detection of HCC showed that the combination of both markers increased the sensitivity from 61% and 74%, for each marker alone, to 91% for both markers combined.19 CT is more accurate than US in identifying HCCs and their lobar or segmental distribution, particularly with the development of helical and multislice spiral scanners. Spiral CT is undertaken without contrast and during arterial (25–50 s), portal (60–65 s) and equilibrium (130–180 s) phases after contrast administration. In addition, it is useful for identifying features of underlying cirrhosis, accurately measuring liver and tumour volumes, and assessing extrahepatic tumour spread. HCCs are usually hypodense and spontaneous hyperdensity is usually associated with iron overload or fatty infiltration, which is seen in 2–20% of patients. Specific features are early uptake of contrast and a mosaic shape pattern. During the portal phase, the density diminishes sharply and results in washout (tumour is hypodense compared to adjacent parenchyma) during the late phase (Fig. 5.3). HCCs may show variable vascularity depending on tumour grade and some are poorly vascularised. The capsule, when present, is best seen during the portal or late phase as an enhanced thickening at the periphery (delayed vascular enhancement is characteristic of fibrosis). Vascular invasion of segmental branches may also be identified. Intratumoral arterioportal fistula may develop and present as early enhancement of portal branches or as a triangular area distal to the tumour with contrast enhancement different from the adjacent parenchyma. Nonetheless, such fistulas are seen frequently in cirrhotic patients without HCC as infracentimetric hypervascular subcapsular lesions. Contrast-enhanced US (CEUS) is the most recent technique to assess vascularisation of tumours. A contrast agent (stabilised microbubbles) is administered intravenously via a bolus injection followed by saline flush. Enhancement patterns are typically described during the arterial (10–20 s postinjection), portal venous (30–80 s) and late phase (120–360 s). Whereas US microbubbles are confined to the vascular spaces, contrast agents for CT and MRI are rapidly cleared from the blood into the extracellular space. The sensitivity of CEUS to detect arterial enhancement is greater than that of CT or MRI because of the continuous monitoring of the images. Washout is slower for well-differentiated than for poorly differentiated tumours. However, it is subject to the same limitations as other US modes: if the baseline scan is unsatisfactory, the CEUS study will also be unsatisfactory. The advent of the second-generation US contrast agent Sonazoid, approved exclusively in Japan in 2007, has made Sonazoid-CEUS more effective for screening and staging than CEUS using other vascular agents such as SonoVue. Sonazoid contrast agent is taken up by Kupffer cells in the postvascular phase or Kupffer phase (starting 10 min postinjection) and provides extremely stable Kupffer images suitable for repeated scanning from 10 to about 120 min after injection.20 Angiography: Although the diagnostic usefulness of angiography has been considerably reduced, it is still widely used as part of arterial chemoembolisation. Arteriography shows early vascular uptake (blush) and, if used, lipiodol injection is retained selectively for a prolonged period by the tumour. On subsequent CT, the retained radiodense lipiodol reveals the tumour as a high-density area. Uptake within the liver is not specific for HCC, since all hypervascular liver tumours, including focal nodular hyperplasia, adenoma, angioma and metastases, will retain Lipiodol. False-negative results may be observed with an avascular, necrotic or fibrotic HCC. Positron emission tomography: The contribution of fluorodeoxyglucose (FDG)-positron emission tomography (PET) in the diagnosis of HCC remains limited because of low sensitivity (60%). FDG-PET can detect poorly differentiated but not-well differentiated HCC due to similarities between the metabolism of FDG in normal hepatocytes and hepatocellular cells. Recently, [18 F]fluorocholine (FCH), a PET tracer of lipid metabolism, has been shown to be significantly more sensitive than [18 F]FDG at detecting HCC, particularly in well-differentiated tumours.21 Interestingly, in metastatic HCC, FDG-PET has a high sensitivity and is more suitable for the detection of bone metastases than CT or bone scintigraphy.22 The first attempt to standardise the diagnostic criteria was in 2000 by the European Association for the Study of Liver Disease (EASLD). Since the last publication of the AASLD practice guidelines for the management of HCC in 2005, several studies have reported that AFP determination lacks adequate sensitivity and specificity for effective surveillance and diagnosis.19 The advocated strategy in the 2011 updated version of the diagnostic criteria23 was based on imaging techniques and/or biopsy as follows: • For nodules less than 1 cm found on US, it was considered that other imaging techniques would be unlikely to reliably confirm the diagnosis. Since the accuracy of liver biopsy for such small lesions and the likelihood of HCC are low, it was felt reasonable to repeat an US at 3- to 6-month intervals until the lesion disappeared, enlarged or displayed characteristics of HCC. If there has been no growth over a period of up to 2 years, routine surveillance can be resumed. • For nodules larger than 1 cm found on US screening of a cirrhotic liver, diagnosis of HCC can be established by one contrast-enhanced imaging technique (multidetector CT or dynamic contrast-enhanced MRI). The specific imaging pattern of HCC is defined by intense contrast uptake during the arterial phase followed by contrast washout during the venous or delayed phases. The value of these non-invasive criteria for HCC in cirrhosis has been confirmed prospectively.24–26 These typical imaging features have a specificity and predictive positive value of approximately 100% and sensitivity of 71%. • If the findings are not characteristic or the vascular features are not typical, and in other clinical settings (e.g. absence of cirrhosis), a diagnostic biopsy was recommended, although it was acknowledged that a negative biopsy did not exclude the diagnosis. Current guidelines23 for the positive diagnosis of HCC are: • For nodules > 1 cm with cirrhosis, early uptake and delayed washout on a single dynamic imaging study (triphasic CT or MRI with gadolinium) is considered characteristic of HCC. • For nodules > 1 cm without cirrhosis or if the vascular features are not typical, a diagnostic biopsy is recommended. • Biopsy of small lesions should be evaluated by expert pathologists. Tissue that is not clearly an HCC should be stained with CD34, CK7, GPC3, HSP70 and GS to improve diagnostic accuracy. • If the biopsy is negative for HCC, it is recommended that the lesion should be re-evaluated using US at 3- to 6-month intervals. If the lesion enlarges but remains atypical for HCC, a repeat biopsy is recommended.
Primary malignant tumours of the liver
Hepatocellular carcinoma
Incidence of HCC
Risk factors for HCC
HBV infection
HCV infection

Human immunodeficiency virus (HIV) infection
Non-alcoholic fatty liver disease (NAFLD)

Hereditary haemochromatosis
Cirrhosis of other aetiologies
Adenoma, contraceptives and androgens
Pathology of HCC and nodular lesions in chronic liver disease
Clinical presentation
Liver function tests and tumour markers
Serum tumour markers
Radiological studies
Computed tomography
Contrast-enhanced ultrasound
Other imaging
Diagnosis of HCC

Primary malignant tumours of the liver