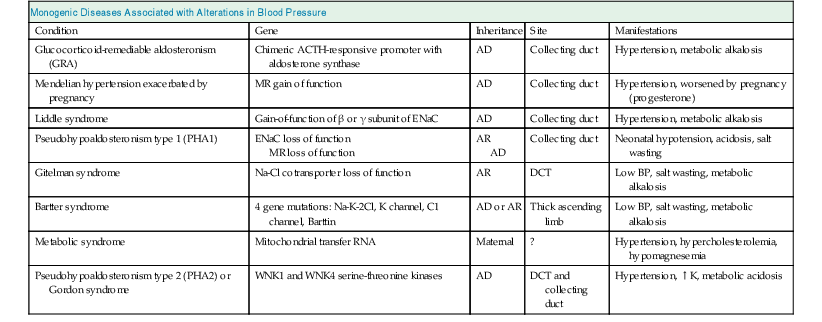
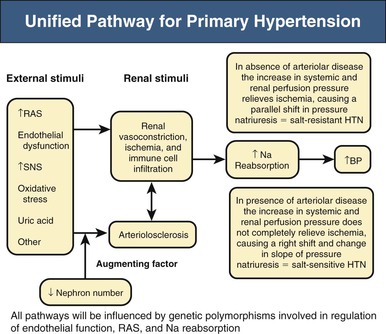
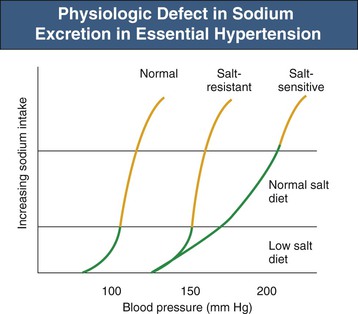
Richard J. Johnson, George L. Bakris, Bernardo Rodriguez-Iturbe Primary (essential) hypertension is defined as blood pressure (BP) greater than 140/90 mm Hg without an identifiable cause. Several readings at different times, following the American Heart Association (AHA) and other guidelines, are necessary to document the diagnosis of primary hypertension. Variability in BP results from several factors, but under normal circumstances, a natural circadian rhythm generates the most significant increase in BP in the morning (6 to 10 am). BP falls during sleep, secondary to a decrease in sympathetic nervous system (SNS) tone and reduced activity of other neuroendocrine systems. There are also minute-to-minute variations in BP (Fig. 34-1). Transient elevations in BP, reaching 150 mm Hg systolic, occur in the majority of normotensive individuals in any given day, especially during exercise.1 However, BP that is 140/90 mm Hg or greater for more than 50% of the day is considered hypertensive. Chapter 33 describes the method and interpretation of BP measurements, including the use of ambulatory BP monitoring. Primary hypertension is classified according to severity (Table 34-1). Stages in hypertension have been adapted by both the Joint National Committee (JNC 7)2 and European Society of Hypertension and European Society of Cardiology (ESH/ESC) guidelines to allow a prognosis to be associated with different levels of BP elevation. The prognosis has been adapted from epidemiologic studies that demonstrate a linear relationship between the risk of cardiovascular events and sustained elevations of arterial pressure. Table 34-1 Classification of blood pressure (JNC 7). (From reference 2.) When only the systolic BP is elevated (systolic BP >140 and diastolic BP <90 mm Hg), the term used is isolated systolic hypertension. The former terms “borderline” or “high-normal” hypertension (defined as BP of 130 to 139/85 to 89 mm Hg) are now classified as prehypertension (defined as BP ≥120 to 139/80 to 89 mm Hg). White coat hypertension is an increase of more than 20 mm Hg in systolic BP noted only in the physician’s office above that seen at home or another setting. In contrast, masked hypertension is BP that is normal in the office but elevated by more than 20 mm Hg when measured by ambulatory BP measurement. Other terms used to describe specific clinical presentations include hypertensive emergencies, associated with acute end-organ damage requiring immediate treatment, usually in a critical care setting, and hypertensive urgencies, when BP needs correction in hours or a few days (see Chapter 37). In hypertensive emergencies, the reduction of BP will halt, prevent, or reverse decreasing glomerular filtration rate (GFR). These terms have replaced “malignant hypertension” and “accelerated hypertension.” Resistant hypertension is defined as hypertension that remains above 140/90 mm Hg despite use of three maximally dosed antihypertensive medications of different classes, including a diuretic. The kidney has a key role in the pathogenesis of hypertension. Guyton and colleagues3 proposed that the kidney in patients with hypertension has a physiologic defect in sodium excretion. Whereas in most individuals an increase in sodium intake is related to an increase in pressure associated with prompt excretion of the salt load, this “pressure-natriuresis” relationship is abnormal in the hypertensive patient (Fig. 34-2). In some hypertensive patients, especially those under age 40, the response to a salt load is similar to that in normal individuals but is shifted rightward, such that higher pressures are required for a specific salt load, called salt-resistant hypertension. In contrast, most hypertensive patients, especially older and African American individuals, have both a rightward shift and a change in the slope, such that BP increases more for the same sodium load, called salt-sensitive hypertension. Additional evidence of a renal defect in sodium handling is the observation that kidney transplantation transfers the susceptibility for hypertension in response to salt in various rat strains. Dietary sodium content also correlates with the prevalence of hypertension in various populations, and intervention studies with salt restriction or loading have shown that the BP response in many hypertensive patients is salt sensitive. The basis for this renal defect in hypertension remains controversial, but three major hypotheses have been proposed. Pickering proposed that hypertension results from the expression of multiple genetic polymorphisms (polygene hypothesis), and Lifton later extended this hypothesis by stating that the cumulative effects of these polymorphisms act to favor sodium retention by the kidney, which would be accentuated in a Westernized society in which there is excessive salt intake (>10 g/day). The observation that numerous monogenic forms of both hypertension and hypotension are mediated by specific mutations involving renal sodium transport, especially involving the epithelial sodium channel (see Chapter 49), supports this hypothesis (Table 34-2). Currently, more than 20 genes have been identified in which mutations or polymorphisms influence blood pressure.4 Many of these involve sodium transport in the distal tubule or the collecting duct. Some heterozygous mutations, such as the Na-K-2Cl cotransporter SLC12A1, the inward rectifier K+ channel KCNJ1 (carrier state for Bartter syndrome), and the heterozygous mutation of the Na-Cl cotransporter SLC12A3 (carrier state for Gitelman syndrome), confer protection from hypertension.5 Whereas genetic polymorphisms influence BP, most studies suggest that genetic mechanisms account for only 20% to 30% of primary hypertension, indicating that other mechanisms are more important in driving the hypertensive response. In 1989, Barker and associates6 reported an increased risk for hypertension in individuals with low birth weight (LBW), and LBW was also found to increase the risk later for diabetes and obesity. Mothers of LBW infants frequently have hypertension, obesity, preeclampsia, or malnutrition, and these maternal factors also carry an increased risk for hypertension in the infant. This has led to interest in the role of “fetal programming” (e.g., from epigenetic changes) in the causation of hypertension. Brenner and colleagues7 have postulated that LBW might lead to hypertension because of impaired renal development, leading to lower nephron number. Maternal malnutrition in laboratory rats predisposes to small babies, low nephron number, and the future predisposition for hypertension. A study of Caucasians with primary hypertension found almost 50% fewer nephrons than in age- and gender-matched controls.8 Studies in African Americans, however, could not confirm the relationship between low nephron number and hypertension. Individuals with LBW also have endothelial dysfunction early in life that likely predisposes to development of hypertension. Some studies suggest this may be caused by uric acid, which is also elevated in LBW infants and children before the development of hypertension. Regardless, LBW cannot be the primary risk factor. For example, one study found that although LBW infants have a 25% risk for developing hypertension as an adult, high-birth-weight infants have a 20% risk.9 Thus, low birth weight and low nephron number likely reflect risk factors for development of hypertension rather than the underlying mechanism. Primary hypertension is frequently associated with renal arteriolosclerosis and hyalinosis with variable degrees of glomerulosclerosis and ischemic tubular injury. Renal function is usually normal or only slightly depressed, whereas renal vascular resistance is high and renal blood flow is low because of intense vasoconstriction of the afferent arteriole. Whereas the microvascular and inflammatory changes may reflect hypertension-associated injury, evidence suggests that these changes may also have a role in the development and maintenance of salt-sensitive hypertension. Salt-sensitive hypertension can be induced by other means that usually involve renal vasoconstriction (Box 34-1). Ischemia stimulates the influx of macrophages and T cells, resulting in oxidative stress, activation of the intrarenal renin-angiotensin system (RAS), and a decrease in local nitric oxide (NO). It is highly likely that the inflammation also activates renal sympathetic nerves. A recent study suggests that rather than responding nonspecifically, the infiltrating T cells may be reacting with specific antigens induced by the local vasoconstriction, particularly heat shock protein 70 (HSP-70).10 Blocking the intrarenal T cell infiltration or infusing T cells that have been tolerized to HSP-70 can block experimental salt-sensitive hypertension in different models.11 Rats born with low nephron number also develop renal microvascular disease and intrarenal T cell accumulation, and administering mycophenolate mofetil (MMF) also reduces BP in these animals. MMF has also been reported to lower blood pressure in hypertensive patients with psoriasis. Renal T cell infiltration is associated with local oxidative stress and angiotensin II (Ang II) generation that results in renal vasoconstriction and an impaired pressure-natriuresis response. This may be augmented by afferent arteriolar remodeling and peritubular capillary loss, which also maintain renal ischemia. Activation of renal afferent sympathetic nerves, likely secondary to the local inflammation, also impairs sodium excretion. The consequence of this condition is a right shift of the pressure-natriuresis curve with a change of the slope, characteristic of salt-sensitive hypertension12 (Fig. 34-3). Although T cells in the kidney play a role in impairing pressure natriuresis, there are also countering mechanisms to reduce the hemodynamic impact of sodium excretion. In particular, the skin can act as a subcutaneous reservoir for sodium as a result of hypertonic accumulation of sodium proteoglycans that activates local macrophages to release vascular endothelial growth factor C (VEGF-C), which stimulates lymphangiogenesis.13 The increase of lymph capillaries attenuates the hemodynamic effects of sodium retention. Depletion of the mononuclear phagocyte system blocks lymphangiogenesis and induces salt-sensitive hypertension. An acute infusion of saline administered to animals with experimentally induced hypertension initially raises blood volume and cardiac output, but the increase in cardiac output is transient and is replaced by a rise in the systemic vascular resistance (SVR). Guyton and colleagues3 called this process “autoregulation,” postulating that the vasoconstriction was an adaptive response resulting from the need to reduce unnecessary increments in tissue perfusion. Several mechanisms may account for the rise in SVR. First, volume expansion increases circulating endogenous cardiotonic steroids, which function as Na+,K+-ATPase inhibitors. For example, ouabain is a digitalis-like factor that is released from the hypothalamus, hippocampus, and pituitary and stimulates the SNS, and marinobufagenin is a digitalis-like factor released from the adrenal cortex. These factors block Na+,K+-ATPase in the kidney, thereby facilitating the excretion of sodium, but at the expense of also blocking Na+,K+-ATPase in the vascular smooth muscle, resulting in increased intracellular calcium with vascular smooth muscle contraction and systemic vasoconstriction.14 Circulating nitric oxide synthase (NOS) inhibitors are also present in some patients with primary hypertension. A small increase in serum sodium concentration from salt ingestion may also stimulate release of vasoconstrictive substances such as vasopressin, which may have a role particularly in African Americans.15 Activation of the SNS also occurs in salt-sensitive hypertension in response to a salt load.16 A possible explanation is that, in the setting of tubulointerstitial injury and intrarenal ischemia, the salt load triggers an intense renal afferent SNS activity that stimulates central nervous system (CNS) sympathetic output.16 The importance of the renal sympathetic nerves in activating the CNS/SNS has become more evident after the introduction of renal artery denervation as a means for treating resistant hypertension (see Chapter 38). Other mechanisms that could contribute to increased SVR are loss of vasodilatory substances from the kidney (e.g., kallikrein, medullipin) or simply the loss of systemic capillaries (microvascular rarefaction).
Primary Hypertension
Definition
Classification of Blood Pressure
BP Classification
Systolic BP (mm Hg)
Diastolic BP (mm Hg)
Normal
<120
and
<80
Prehypertension
120-139
or
80-89
Stage 1 hypertension
140-159
or
90-99
Stage 2 hypertension
≥160
or
≥100

Etiology and Pathogenesis
Genetic (Polygene) Hypothesis
Congenital (Low Nephron Number) Hypothesis
Acquired Renal Injury Hypothesis
Role of Renal Injury in Sodium Retention
How Does Salt Retention Lead to Hypertension?
Pathogenic Mechanisms Driving the Current Epidemic of Hypertension
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Primary Hypertension
Chapter 34
Figure 34-2 Physiologic defect in sodium excretion in primary hypertension. Evidence suggests that in patients with primary hypertension, a higher blood pressure is required to excrete an individual sodium load. In salt-resistant hypertension, the pressure-natriuresis curve has a rightward but parallel shift; with salt-sensitive hypertension, it is both a shift to the right and a change in slope. (Modified from reference 4.)