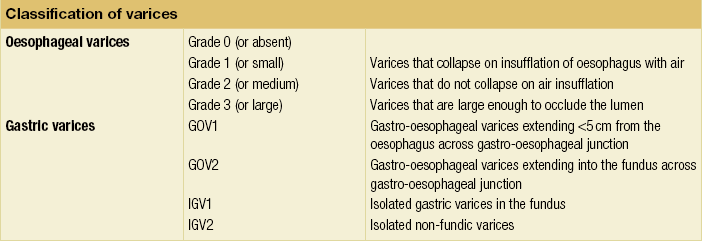
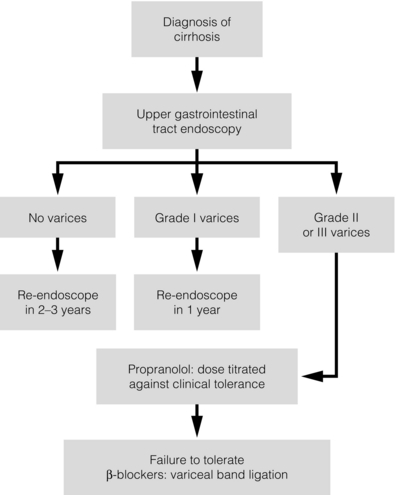
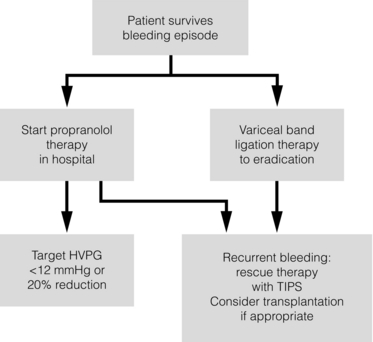
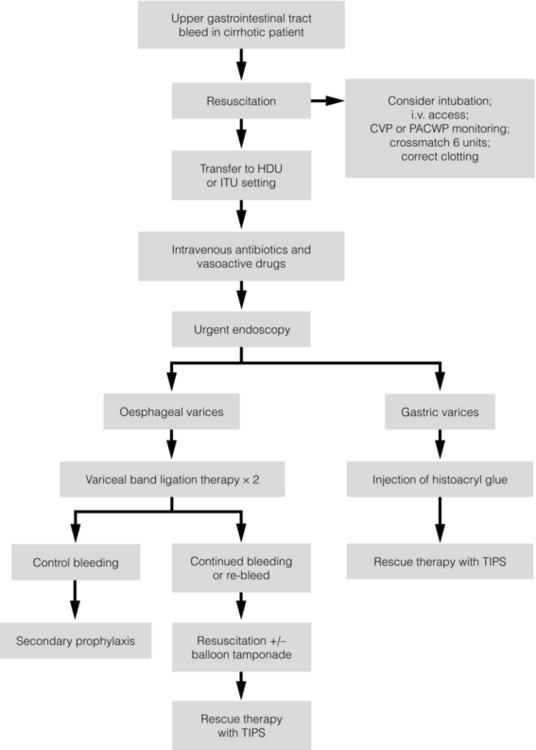
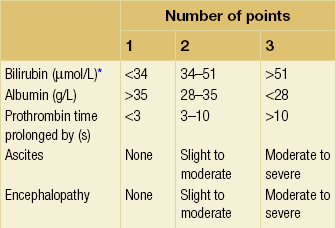
8 Traditionally, portal hypertension has been classified as prehepatic, intrahepatic or posthepatic, with the intrahepatic causes subdivided into presinusoidal, sinusoidal and postsinusoidal (Table 8.1). Prehepatic causes are usually due to portal vein thrombosis, which is discussed later in this chapter. The main cause of portal hypertension in the West is cirrhosis. This is a sinusoidal obstruction to portal flow with varying causes. Viral hepatitis and alcoholic liver disease are the most common causes, but others include primary biliary cirrhosis, primary sclerosing cholangitis and haemochromatosis. Presinusoidal obstruction due to hepatic fibrosis occurs in schistosomiasis. Worldwide, this is one of the commonest causes of portal hypertension and, as it is usually associated with normal liver function, has a better prognosis. The main causes of postsinusoidal portal hypertension are hepatic venous thrombosis (Budd–Chiari syndrome) and veno-occlusive disease. Table 8.1 Experimental studies have demonstrated that the initial factor in the pathophysiology of portal hypertension is the increase in vascular resistance to portal blood flow. In cirrhosis, this increase in resistance occurs in the hepatic microcirculation (sinusoidal portal hypertension), and is a consequence of both a ‘passive’ and an ‘active’ component. The ‘passive’ component is the mechanical consequence of the hepatic architectural disorder resulting from histological cirrhosis, and the ‘active’ component is the active contraction of portal/septal myofibroblasts, activated stellate cells and portal venules. The increase in intrahepatic tone is probably a consequence of an imbalance between an increase in the endogenous vasoconstrictor substances, such as endothelin, noradrenaline, leukotrienes and angiotensin II, and a relative decrease in the endogenous vasodilator nitric oxide.1 Vasodilatory drugs (for example, calcium channel blockers) may restore the equilibrium in intrahepatic tone, although they are not used for this indication in clinical practice. The prevalence of oesophageal varices in patients with cirrhosis and portal hypertension is high. When cirrhosis is diagnosed, varices are present in 40% of compensated and 60% of decompensated cirrhotics.2 After the initial diagnosis of cirrhosis, varices develop with an incidence of 5% per year; subsequently, they may progress from small to large at an incidence of 10–15% per year.3 Rapid progression of hepatic decompensation is associated with a rapid increase in size, whilst improvement in liver function, particularly when associated with removal of the injurious agent (e.g. abstinence from alcohol), may result in a decrease in size or disappearance of the varices.4,5 The overall incidence of variceal bleeding following diagnosis is of the order of 25% in unselected patients. The most important predictive factors of variceal bleeding are severity of liver dysfunction, size of varices and intravariceal wall pressure (which although difficult to measure may correlate at endoscopy with the presence of red spots or red weals).6 Traditionally, liver dysfunction has been classified using the Child–Pugh score7 (Table 8.2), but a more recent scoring system, the MELD (Model for End-stage Liver Disease), may be a better prognostic indicator (Box 8.1).8 Variceal size may be the best single predictor of variceal bleeding and generally it is used to decide whether a patient should be given prophylactic therapy or not. Whether a patient dies from a variceal bleed depends on the severity of the accompanying liver failure; those with a high Child–Pugh or MELD score have been reported to have as high a risk of mortality as 30–50% within 6 weeks of the index bleed.9 However, a more realistic figure would be 20% at 6 weeks with an immediate mortality from uncontrolled bleeding as low as 5–8%. Indeed, in 40–50% of patients who bleed and develop hypotension, variceal bleeding stops spontaneously, probably as a result of reflex splanchnic vasoconstriction with associated reduction in portal pressure and blood flow; this beneficial response is nullified by over-transfusing the patient. Box 8.1 Model for End-stage Liver Disease (MELD) MELD is calculated for patients over the age of 12 based on the following variables: The formula incorporates these variables as: The following rules must be observed when using this formula: • 1 is the minimum acceptable value for any of the three variables. • The maximum acceptable value for serum creatinine is 4. • The maximum value for the MELD score is 40. All values higher than 40 are given a score of 40. • If the patient has been dialysed twice within the last 7 days, then the value for serum creatinine used should be 4.0. Table 8.2 Grade A 5–6 points; Grade B 7–9 points; Grade C 10–15 points. *In primary biliary cirrhosis, the point scoring for bilirubin level is adjusted as follows: 1, < 68; 2, 68–170; 3, > 170. The incidence of re-bleeding ranges between 30% and 40% within the first 6 weeks; this risk peaks in the first 5 days following the index bleed. Bleeding gastric varices, active bleeding at emergency endoscopy, low serum albumin levels, renal failure and a hepatic venous pressure gradient > 20 mmHg have all been reported as significant indicators of an early risk of re-bleeding.10–12 Patients surviving a first episode of variceal bleeding have a very high risk of re-bleeding (63%) and death (33%), and this is the basis for treating all patients to prevent further bleeding.9 Portal hypertension may present acutely with variceal bleeding or be discovered during the investigation of a patient with liver disease. Varices are usually easily diagnosed at endoscopy and patients will then be investigated systematically. A classification of the grading of varices is given in Table 8.3. Presentation of patients with liver disease is variable and ranges from non-specific tiredness to advanced encephalopthy with decompensation. External features of advanced liver disease such as spider naevi, palmar erythema and ascites are easy to detect, although these signs will be lacking in many patients. Splenomegaly is probably the most useful physical sign, although some patients will have the classic sign of dilated umbilical vein collaterals (caput medusae). Varices do not develop until the HVPG increases to 10–12 mmHg and the HVPG must be greater than 12 mmHg for the appearance of complications such as variceal bleeding and ascites.13 Longitudinal studies of patients with complications of portal hypertension have demonstrated that when an HVPG decreases to less than 12 mmHg with pharmacological therapy, TIPS or an improvement in liver function, variceal bleeding is prevented and varices may decrease in size or disappear altogether.14 When this target is not reached, a substantial reduction in portal pressure by more than 20% still offers protection against variceal bleeding15 and thus these two parameters are regarded as the end-points to therapeutic strategies to lower portal pressure. Recent evidence suggests that these therapeutic end-points may also reduce the risk of other complications of portal hypertension, including ascites, spontaneous bacterial peritonitis and hepatorenal syndrome.16,17 Primary prophylaxis for the prevention of variceal haemorrhage A meta-analysis has indicated that indefinite treatment with propanolol or nadolol significantly reduces the bleeding risk from 25% with non-active treatment or placebo to 15% with beta-blockers over a median follow-up period of 24 months; there was no significant reduction in mortality.3 The benefit of therapy was only proven in those patients with grade II (or larger) varices; there was no evidence to support the use of primary prophylactic therapy in patients with grade I varices. Withdrawal of therapy was associated with a return to the same bleeding risk (25%) as the untreated subpopulation; indeed, there may also be an increased risk of mortality over untreated patients in those individuals who stop therapy.18,19 Assessment of the success of primary prophylactic therapy is ideally undertaken by measurement of the HVPG before and after initiating therapy, with the aim being to reduce the HVPG to < 12 mmHg or to reduce it by > 20% from its baseline value.14 In practice, however, measurement of HVPG does require specific training and is probably not cost-effective for assessing primary prophylactic therapy. Thus, the clinician faces the question of how to adjust the dose of beta-blocker to maximise its beneficial effects. Traditional practice has recommended a stepwise increase in dose until the heart rate decreases by 25%, is < 55 beats per minute, or there is arterial hypotension or clinical intolerance. This means that the dose of the beta-blocker is titrated against its β1 effects (cardiac) and clinical tolerance; however, a fall in portal pressure results from blockade of both β1 and β2 receptors, and the fall in portal pressure does not readily correlate with the fall in heart rate or blood pressure. Therefore, titration solely against clinical tolerance may be the most useful surrogate marker of the maximal dose of beta-blocker in the absence of HVPG measurement. When compared with propanolol, carvedilol significantly increased the number of patients achieving a target reduction of HVPG (< 12 mmHg or > 20% reduction from baseline HVPG).19 There is considerable controversy about how to give the carvedilol because of its hypotensive side-effects; however, the above study demonstrated that lower doses (12.5 mg/day) result in good tolerance. In practice, the usual starting dose is 6.25 mg/day and the usual maintenance dose 12.5 mg/day. In patients who are unable to tolerate beta-blockers (15–20%) because of side-effects or relative/absolute contraindications (for example, asthma), treatment with nitrates is ineffective, despite its portal pressure-lowering properties.20 Therefore, variceal band ligation (VBL) therapy is the only option for patients with high-risk varices (grade II or above) and contraindications to beta-blockers. More controversially, a meta-analysis has suggested that VBL is a more effective mode of treatment than beta-blockers for primary prophylaxis.21 However, this analysis included four trials, only two of which have been published in full; therefore, it seems reasonable to recommend that, for the time being, beta-blockers remain the primary prophylactic therapy of choice in terms of cost and convenience. Of course, VBL does not reduce portal pressure (and therefore measurement of HVPG following endoscopic monotherapy is of no value) and this may leave the patient at risk of developing other complications of portal hypertension. An algorithm for the primary prevention of variceal bleeding is given in Fig. 8.1. Following a variceal bleed, patients with cirrhosis should be managed in two ways: firstly, they should receive urgent and active treatment for the prevention of re-bleeding; secondly, they should be examined for signs of physiological stress following their bleed, which might indicate a need for an elective liver transplant assessment (Fig. 8.2). Management of non-cirrhotic patients is discussed later in this chapter. Meta-analyses of studies using beta-blocker therapy to prevent re-bleeding have demonstrated both a significantly decreased mortality (27% in controls to 20% in beta-blocker-treated individuals) and a decreased incidence of re-bleeding (63% to 42%).3 VBL also both improves survival and significantly decreases re-bleeding rates; it is superior to endoscopic sclerotherapy since it is associated with significantly fewer complications.22,23 Currently, it is unclear whether pharmacological therapy is better than VBL or vice versa; studies have demonstrated a variety of outcomes with reference to re-bleeding rates, but none have indicated any clear difference in survival.15,24,25 A combination of pharmacological therapy and endoscopic therapy is commonly used, but evidence suggesting a better outcome with this combination compared with monotherapy is hard to find. Likewise, combination therapy of nitrates and beta-blockers has not been consistently shown to be more effective than beta-blockers alone or VBL.15,26 Antibiotics should be instituted from admission, since these increase the survival of bleeding patients; norfloxacin 400 mg/12 hours or ciprofloxacin 250 mg/12 hours are the antibiotics of choice.27,28 Finally, early therapy should also involve starting a vasoactive drug from admission (usually terlipressin or octreotide); a number of randomised controlled trials demonstrate that early administration of vasoactive drugs facilitates endoscopy, improves control of bleeding and reduces the 5 day re-bleeding rate.29,30 Initiation of these measures in association with endoscopic therapy at the time of diagnostic endoscopy will control bleeding in approximately 75% of patients. However, as in most trials, in acute variceal bleeding this combined approach failed to improve overall mortality compared with drug or endoscopic therapy alone. The optimal duration of vasoactive drug therapy is not well established and requires evaluation; current recommendations are to continue the drug for 5 days, since this covers the period of maximum risk of re-bleeding. Endoscopic therapy should be performed at the time of diagnostic endoscopy, within 12 hours of admission in a resuscitated patient. However, if the patient is stable, endoscopic therapy can probably be postponed until within normal working hours. There are multiple randomised controlled trials examining modes of endoscopic therapy in acute variceal bleeding. These have compared: endoscopic therapy with no therapy; endoscopic therapy with vasoactive drug therapy; endoscopic sclerotherapy with variceal band ligation therapy; combined endoscopic therapy with variceal band ligation therapy; and endoscopic therapy with TIPS. Endoscopic therapy is certainly superior to no therapy;31 of the two endoscopic therapies, variceal band ligation therapy should be considered the treatment of choice since it is associated with significantly fewer complications (oesophageal stricturing or oesophageal ulcer formation) and significantly fewer sessions of therapy to eradicate the varices. However, there is probably no difference in re-bleeding or mortality rates between the two therapies. Likewise, there is little evidence to support combined endoscopic therapy for the treatment of bleeding varices.32 In practice, however, it is sometimes beneficial for the endoscopist to use a small volume of sclerosant initially to improve vision in order to place some variceal bands to achieve eventual haemostasis. If endoscopic therapy fails to control bleeding, balloon tamponade should be used as a ‘bridge’ until definitive therapy can be offered. In practice, this usually means a further attempt at endoscopic band ligation therapy followed by second-line therapies. An algorithm for the management of variceal bleeding in cirrhotics is given in Fig. 8.3.
Portal hypertension
Aetiology and pathophysiology of portal hypertension
Presinusoidal
Sinusoidal
Postsinusoidal
Extrahepatic
Cirrhotic
Budd–Chiari syndrome
Portal vein thrombosis
Postviral (B, C)
Veno-occlusive disease
Splenic vein thrombosis
Alcoholic
Caval web
Increased splenic flow (tropical splenomegaly, myelofibrosis)
Cryptogenic
Primary biliary cirrhosis
Primary sclerosing cholangitis
Constrictive pericarditis
Intrahepatic
Chronic active hepatitis
Schistosomiasis
Haemochromotosis
Congenital hepatic fibrosis
Wilson’s disease
Sarcoidosis
Non-cirrhotic
Acute alcoholic hepatitis
Cytotoxic drugs
The natural history of portal hypertension
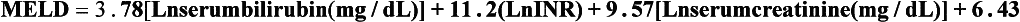
Presentation
Therapeutic aims for pharmacological therapy in portal hypertension

Oesophageal varices


Prevention of re-bleeding from oesophageal varices (secondary prophylaxis)

Treatment for bleeding oesophageal varices