(1)
Al Agouza, Cairo, PO, Egypt
2.1 Embryology and Penile Development
Between the fourth and seventh weeks, the mesodermal mesenchyme migrates to the cranial aspect of the cloacal membrane to form the genital tubercle. The cloacal membrane itself is composed of two layers: endoderm and ectoderm. The caudal portion of the cloacal membrane develops into urogenital folds. These structures are the precursors for external genitalia in both males and females. In those with a Y chromosome, the SRY gene signals the differentiation of primitive sex cords into the testes by first signaling the development of Sertoli cells. Sertoli cells then aid in the development of germ cells and Leydig cells within the testes. Leydig cells produce testosterone, which is converted to dihydrotestosterone to induce external genitalia development. Normal embryogenesis of the male genitalia involves the formation of the penis and scrotum. The early development of the external genitalia in the two sexes is similar before the ninth week of gestation. Understanding factors and sequential steps in normal embryogenesis is fundamental in the comprehension of the pathogenesis of male genital anomalies. These factors include testosterone synthesis by the fetal testis and its enzymatic conversion into dihydrotestosterone by 5α-reductase and the presence of androgen receptors able to recognize the androgenic hormones. The influence of dihydrotestosterone on the androgen receptors results in the differentiation of the genital tubercle, genital (labioscrotal) folds, and genital swelling between 9 and 13 weeks of gestation into the male structures of the glans penis, penile shaft, and scrotum, respectively [1].
The male develops in a proximal to distal manner. As the penis forms from the elongation and enlargement of the phallus, the lateral walls of the urethral groove form from the ventrally located genital folds. The genital folds then fuse in the midline. The glanular urethra forms from the ingrowth of surface epithelium, but this long-held theory has been challenged with evidence suggesting that it is due to the fusion of the urethral plate. The scrotum forms through the inferomedial migration and midline fusion of the genital folds as delineated by the scrotal raphe. In females and in males with abnormalities in testosterone and/or dihydrotestosterone production, 5α-reductase deficiency, or androgen receptor insufficiency, the genital tubercle, genital folds, and genital swellings passively become the clitoris, labia minora, and labia majora, respectively.
Congenital penile or scrotal anomalies can be isolated variations of external genitalia development, or they can represent significant underlying malformations. Prompt diagnosis and potential surgical planning are essential to allay the anxieties of parents as well as to identify other potentially clinically significant conditions. Congenital penile anomalies have been shown to be increasing in incidence. The weighted incidence of anomalies including hypospadias has increased from 7 of every 1,000 newborns (1988–1991) to 8.3 of every 1,000 newborns (1997–2000), but this may be a result of increased reporting. Multiple etiologies in abnormal development have been proposed, including genetic, hormonal, and environmental influences. Whereas molecular pathology has aided in the identification of key steps in abnormal development, modern imaging techniques continue to refine the evaluation and treatment of such anomalies [2].
2.2 Penile Agenesis (PA)
Nomenclature
Aphallia, apenia, ablatio penis, penile agenesis
Definition
Congenital absence of the penis, which is a rare anomaly caused by developmental failure of the genital tubercle
Historical Background
The earliest case report of aphallia was by Imminger in 1853; since then approximately 80 cases have been reported in the literature, but very recently many cases in the process of reporting [3].
Approximate Incidence
1 in 10–30 million population, the incidence of stillbirth or neonatal death is approximately one-third of cases.
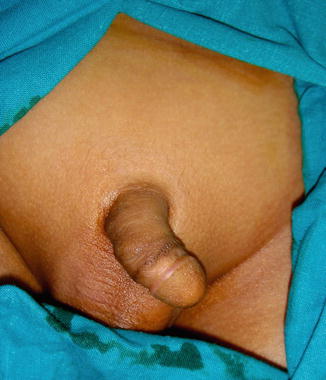
The phallus is completely absent, including the corpora cavernosa and corpus spongiosum. Usually, the scrotum is normal and the testes are undescended, but there are many cases reported where both testicles are normally descended with normal development (Figs. 2.1 and 2.2).
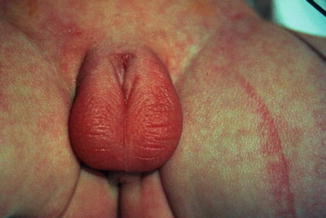
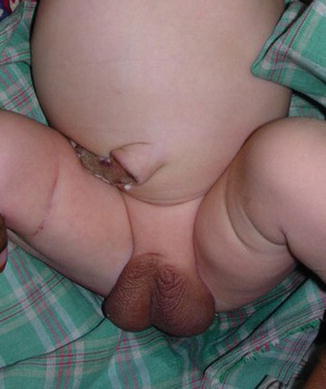
Fig. 2.1
Complete aphallia with normal scrotum and testicles
Fig. 2.2
Neonate with aphallia and normal testicle
The urethra opens at any point of the perineal midline from over the pubis to, most frequently, the anus or anterior wall of the rectum. In Fig. 2.3, no urethral opening could be appreciated.
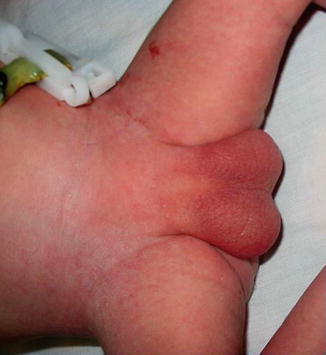
Fig. 2.3
Aphallia without urethral opening
Etiology
Agenesis of the penis occurs as a consequence of single gene disorders, teratogenic effects, or malformation sequences and associations of unrecognized patterns of anomalies. It thus should be considered a developmental field defect. Its concurrence with scrotal hypoplasia, absent raphe, and anal anomalies implies a major disturbance of the caudal mesoderm; in such cases severe renal defects are usually seen, and the prognosis is poor. When the patient has a patent urethra and normal scrotum, raphe, and testes, the baby may survive with such anomaly; however, penile agenesis may be a localized malformation of the genital tubercle potentially related to penoscrotal transposition. Reports indicate that aphallia may be associated with pregnancy complicated by poorly controlled maternal diabetes [4].
Associated Anomalies
More than 50 % of patients with penile agenesis have associated genitourinary anomalies, the most common of which is cryptorchidism, renal agenesis, and dysplasia. Cardiovascular gastrointestinal defects, such as caudal axis anomalies, also have been described. Skoog and Belman [5] reviewed 60 reports of aphallia and found that the more proximal the urethral meatus, the greater the likelihood of neonatal death and the higher the incidence of other anomalies. Sixty percent of patients had a postsphincteric meatus located on a peculiar appendage at the anal verge. This group of patients had the highest survival rate (87 %) and the lowest incidence of other anomalies (1.2 per patient). Twenty-eight percent of patients had presphincteric urethral communications (prostatorectal fistula), and there was a 36 % neonatal mortality rate. Twelve percent had urethral atresia and a vesicorectal fistula for drainage. This group had the highest incidence of other anomalies and an almost a 100 % mortality rate (Fig. 2.4).
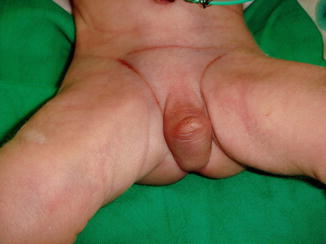
Fig. 2.4
Aphallia with absent testicles and only skin covering the urethral opening
Classifications
Skoog and Belman [5] suggested three variants, based on urethral position in relationship to the anal sphincter, as postsphincteric, presphincteric (prostatorectal fistula), and urethral atresia. The more proximal the bladder outlet, the greater the likelihood of other anomalies and death.
We adopted herein and after reviewing many cases and literature concerning with this issue another classification according to presence or absence of external urinary meatus and if this problem takes place with another syndrome or not:
Aphallia with other genitourinary anomalies
Aphallia with other syndromes
Aphallia with absent urethra
Aphallia with caudal regression syndrome
All reported cases of aphallia with absent external urinary meatus showed short span of life and there is no record of any survival whatever the measurers taken, and most of those cases are associated with imperforate anus and showing a degree of caudal regression (Fig. 2.5), and this could explain the association between aphallia and sirenomelia (Fig. 2.6). This classification correlated with Evans et al. who suggest that most cases can be classified into either a severe form (16 %) with renal aplasia or dysplasia and other caudal anomalies or a second group (72 %) with low mortality and fewer additional malformations [6].
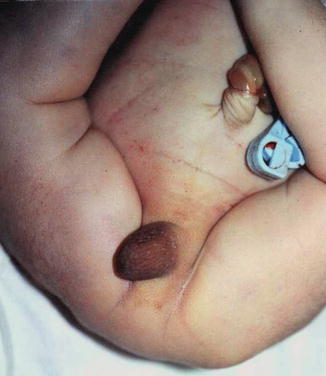
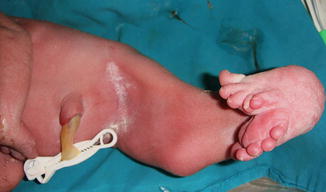
Fig. 2.5
Aphallia with partial caudal regression, no anal opening, and a hump of skin replacing the phallus
Fig. 2.6
Aphallia in association with sirenomelia
Diagnosis
The diagnosis of PA requires the absence of corpora cavernosa and corpus spongiosum with urethral opening at any point on the perineum in midline, over the pubis, at the anterior aspect of the scrotum, or, most frequently, just anterior to the anus and anterior wall of the rectum. This rare entity should be differentiated from concealed penis, rudimentary penis, micropenis, epispadias, hypospadias pseudohermaphroditism, and intrauterine amputation of the penis. Anorectal anomalies such as imperforate anus, congenital rectal strictures, and rectovesical fistula, cryptorchid testis, hydrocele, hernia, renal dysplasia, horseshoe kidneys, and agenesis of the prostate could be an associated malformations [7]. When oligohydramnios or anhydramnios hinders proper diagnosis by ultrasound, MRI is an excellent technique for revealing the anatomy of genitourinary anomalies in the fetus, and many cases of aphallia could be diagnosed early in pregnancy along with other associated anomalies [8].
Management
Children with this lesion should be evaluated immediately with a karyotype and other appropriate studies to determine whether there are associated malformations of the urinary tract or other organ systems. Gender reassignment was recommended for affected newborns in the past. However, with more recent revelations that some of these patients have a male gender identity despite reconstruction as a female, the recommendation to perform gender reassignment should be made very carefully and only after full evaluation by an ambiguous genitalia assessment team that includes a pediatric urologist, endocrinologist, and psychiatrist. As a male, the patient would potentially be fertile, but currently there is an inability to construct a cosmetically acceptable phallus that would allow normal urinary, sexual, and reproductive function. The issues are similar to those under consideration in many genetic males born with cloacal exstrophy. Gender reassignment involves orchiectomy and feminizing genitoplasty in the newborn period. At a later age, construction of a neovagina is necessary. Urinary tract reconstruction with simultaneous construction of an intestinal neovagina through a posterior sagittal and abdominal approach in patients with penile agenesis has been described [9].
Infants with penile agenesis historically have undergone gender reassignment surgery, including bilateral orchiectomy with preservation of the scrotal skin for later vaginal reconstruction, labial construction, and urethral transposition [10]. However, questions remain regarding in utero gender imprinting and the long-term psychological effects of gender conversion, and increasing controversy surrounds the timing, role, and the necessity of gender reassignment. The long-accepted notion regarding the presence of a phallus or phenotypic phallic growth potential should not be the major criterion in recommending gender reassignment.
2.3 Microphallus
Nomenclature
Micropenis
Definition
The term microphallus, or micropenis, is applicable only to a normally formed yet abnormally short penis. The term specifically applies to a penis with a stretched length more than 2.5 standard deviations (SD) less than the mean for age (Table 2.1). In general, the penis of a full-term neonate should be at least 1.9 cm long. One must differentiate buried penis or webbed penis from the micropenis, with the former having a normal penile shaft. Measurement (stretched penile length) is very important in differentiation of the various types of pseudomicropenis, particularly the buried penis in the obese infant and the penis concealed by an abnormal skin attachment or excessive suprapubic fat which is commonly referred to as an inconspicuous penis.
Age | Mean ± SD | Mean-SD |
|---|---|---|
Newborn 30-week gestation | 2.5 ± 0.4 | 1.5 |
Newborn 34-week gestation | 3 ± 0.4 | 2.0 |
0–5 months | 3.9 ± 0.8 | 1.9 |
6–12 months | 4.3 ± 0.8 | 2.3 |
1–2 years | 4.7 ± 0.8 | 2.6 |
2–3 years | 5. 1 ± 0.9 | 2.9 |
3–4 years | 5.5 ± 0.9 | 3.3 |
4–5 years | 5.7 ± 0.9 | 3.5 |
5–6 years | 6 ± 0.9 | 3.8 |
6–7 years | 6.1 ± 0.9 | 3.9 |
7–8 years | 6.2 ± 1.0 | 3.7 |
8–9 years | 6.3 ± 1.0 | 3.8 |
9–10 years | 6.3 ± 1.0 | 3.8 |
10–11 years | 6.4 ± 1.1 | 3.7 |
Adult | 13.3 ± 1.6 | 9.3 |
This condition may be considered a minor form of ambiguous genitalia with correlated medical and psychological problems similar to those of the major intersex form. The scrotum usually is normal (Fig. 2.7), but sometimes the testes are small or undescended, or the scrotum may migrate cephalically engulfing the small phallus as a minimal scrotal transposition (Fig. 2.8). In a few cases, the corpora cavernosa are severely hypoplastic, and it is not rare to have microphallus with severe hypospadias and deficient corpus spongiosum (Fig. 2.9).
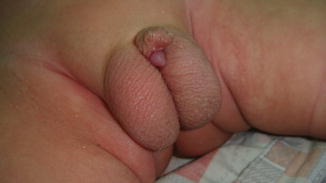

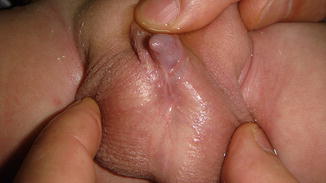
Fig. 2.7
Microphallus with normal testicles
Fig. 2.8
Microphallus with scrotal transposition
Fig. 2.9
Microphallus with corpus spongiosum deficiency and hypospadias
Historical Note
Perceptions of penile size are culture specific, so in ancient Greece and in Renaissance art, an uncircumcised and small penis was culturally seen as desirable in a man, whereas a bigger or circumcised penis was viewed as comical or grotesque. Ancient Rome may have had a contrary view, and a larger penile size was preferred in medieval Arabic literature.
Incidence
The condition is thought to affect 1 in 200 males that are born. According to the Network on Psychosexual Differentiation, incidence for a micropenis is below 2 %. In Colombia, the incidence is 19:100,000 people, while the incidence for hypospadias in the same study, a factor of 10 higher [12].
The observed significant increase in recent years of such cases in neonates by some authors is probably due to the influence of exogenous substances, such as antiandrogens, estrogen, and chemical compounds that bind to the androgen receptors [13].
Etiology
Micropenis results from a multiplicity of endocrine and nonendocrine conditions. The most common etiologies include hypogonadotropic hypogonadism, hypergonadotropic hypogonadism, and idiopathic micropenis [14].
In hypogonadotropic hypogonadism, secretion of gonadotropin-releasing hormone (GnRH) by the hypothalamus is impaired. This leads to decreased pituitary secretion of luteinizing hormone and follicle-stimulating hormone, depriving the testis of its stimulus to secrete testosterone. This pathogenesis exists in some hypothalamic dysfunctions, such as Kallmann syndrome or Prader–Willi syndrome.
Micropenis secondary to hypergonadotropic hypogonadism is associated with conditions in which the testes are impaired functionally and unable to respond to hypothalamic–pituitary stimulation; an example of this condition is gonadal dysgenesis.
In so-called idiopathic micropenis, endocrine analysis demonstrates a normal hypothalamic–pituitary–testicular axis, but some recognized causes could be implicated:
Primary testicular failure, e.g., anorchia, partial gonadal dysgenesis, and Klinefelter’s syndrome
Hypogonadotropic hypogonadism, Kallmann syndrome, and CHARGE syndrome
Defects in testosterone action, partial androgen insensitivity, and 5α-reductase deficiency
Developmental anomalies, aphallia, and cloacal exstrophy [15]
Differential Diagnosis
Congenital micropenis should be differentiated from:
Inconspicuous penis, which refers to a penis that appears to be small with a normal stretched penile length measured from the pubic symphysis to the tip of the glans and normal diameter of the penile shaft.
Buried penis, also referred to as hidden or concealed penis, is a form of inconspicuous penis. A buried penis is a normally a developed penis that is hidden away by the suprapubic fat pad.
Webbed penis
Management
Topical application of 5 % testosterone cream causes increased penile growth. The objective is to provide sufficient testosterone to stimulate penile growth without altering growth and closure of the epiphyses. Therapy should be started by age 1 year and aimed at maintaining genital growth commensurate with general body growth. Hormonal stimulation, especially with dihydrotestosterone, may produce some penile growth even after puberty. This can be given in a 2.5 % gel formulation once per day, with review after 6–8 weeks to assess the effect. The most common therapeutic regimen for injectable testosterone is testosterone enanthate 25–50 mg intramuscularly once a month for 3 months [16].
Patients with micropenis who also suffer from penile dysmorphic disorder require careful and intensive psychological counseling. Corrective surgery for micropenis can be performed in patients with realistic expectations. Total phalloplasty using radial artery-based forearm skin flaps can offer restoration of normal penile length in selected patients. More conservative surgical techniques to improve length or girth are limited by minimal enhancement but associated with a significantly lower rate of complications and comorbidity compared to total phalloplasty. Emerging tissue engineering techniques might represent a suitable alternative to penile replacement surgery in the future [17].
Because micropenis is the result of numerous pathological conditions, assignment of sex of rearing generally is deferred until a physician determines whether the penis can grow in response to testosterone administration or not. In individuals with microphallus who are insensitive to the androgen, castration and gender conversion can be considered. However, in most patients with micropenis, male gender assessment can be maintained with androgen stimulation [18].
2.4 Megalopenis
Definition
Abnormal largeness of the penis is an anomaly whereby the phallus enlarges rapidly in childhood due to high level of production of testosterone, e.g., interstitial cell tumors of the testicle, hyperplasia or tumors of the adrenal cortex, or secondary to other congenital anomalies.
Nomenclature
Megalopenis, macrophallus, and macropenis
Etiology
Hypothalamic tumor associated with precocious puberty may be the cause of macrophallus. Benign familial infantile seizures with inversion of chromosome 15 are reported to be associated with macrophallus, and also some cases are reported with heterochrony development where deletion of chromosomal region 13q21q31 is associated with macropenis [19].
Femoral hypoplasia–unusual facies syndrome (FHUFS) which is characterized by bilateral, mostly asymmetrical, femoral hypoplasia with variable lower limb shortening and nonspecific facial dysmorphism is commonly associated with megalopenis [20].
Macropenis is an uncommon finding of Fraser syndrome, which is characterized by cryptophthalmos, cutaneous syndactyly, malformations of the larynx and genitourinary tract, craniofacial dysmorphism, orofacial clefting, mental retardation, and musculoskeletal anomalies. The inheritance is autosomal recessive. No diagnostic cytogenetic abnormalities have been documented in affected patients, and no molecular genetic studies have been reported.
Isolated cases of macropenis in a normal baby and without any associated anomalies are extremely rare (Fig. 2.10), but macropenis could be noticed in other congenital penile or urethral anomalies which will be discussed later on like congenital megalourethra (Fig. 2.11) and congenital penile lymphedema (Fig. 2.12); in those conditions the penile gigantism is not a true one. Megalopenis should be also differentiated from megaprepuce or macroposthia which also will be discussed in Chap. 3.
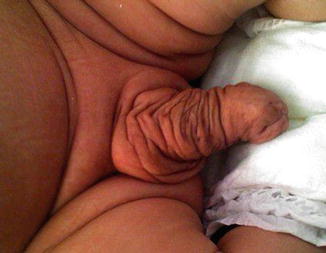
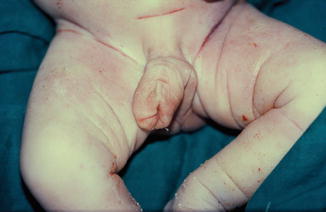

Fig. 2.10
Isolated megalopenis
Fig. 2.11
Megalopenis in congenital megalourethra
Fig. 2.12
Megalopenis in congenital lymphedema
2.5 Penile Duplication (PD)
Definition
Duplication of the penis, or diphallia, is a rare anomaly resulting from incomplete fusion of the genital tubercle. It is an extremely rare but well-documented anomaly.
Nomenclature
Diphallia, penile duplication, diphallic terata, or diphallasparatus
Incidence
It is estimated to occur in one out of five million live births. It is usually accompanied by other congenital anomalies such as renal, vertebral, hindgut, or anorectal duplication; also there is a higher risk of spina bifida.
Historical Background
The first reported case of PD was reported by Johannes Jacob Wecker in 1609 [21]. Penile duplication is a normal finding in some animal species, male snakes and lizards, each possessing a pair of penis-like organs.
Definition
Duplication of the penis is a rare anomaly and has a range of appearances from a small accessory penis to complete duplication. In some cases, each phallus has only one corporal body and urethra, whereas others seem to be a variant of twinning, with each phallus having two corpora cavernosa and a urethra (Fig. 2.13). The penises usually are unequal in size and lie side by side, but very rarely the other moiety lies beneath the first one in a sagittal plane (Fig. 2.14).


Fig. 2.13
Complete diphallia with two urethral openings and two scrotums
Fig. 2.14
Diphallia in a sagittal plane
Associated anomalies are common, including hypospadias, bifid scrotum, and duplication of the bladder, renal agenesis or ectopia, and diastasis of the pubic symphysis. Anal and cardiac anomalies also are common (Fig. 2.15). Evaluation should include imaging of the entire urinary tract. Sonography has been reported to aid in assessment of the extent of phallic development [22]. MRI can also be used to assess penile development. MRI is a valuable method for achieving the accurate diagnosis of these anomalies and associated malformations. It also provides the appropriate knowledge regarding anatomical detail and assists the surgeon in decision making and preoperative planning for the optimal surgical approach [22].
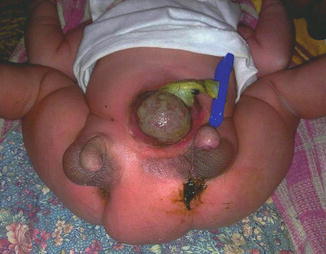
Fig. 2.15
Complete diphallia with two phalli and two anal openings and exomphalos
Etiology
Hollowell et al. [23] reviewed the embryogenesis of diphallia and suggested that complete diphallus could result from longitudinal duplication of the infraumbilical cloacal membrane before the fourth week of gestation, the subsequent mesodermal migration allowing two separate, complete sets of genital tubercle, genital folds, and genital swellings to develop. The fusion of the genital folds and swellings may not, however, be entirely normal, accounting for the finding that one of the two urethras may be a blind pit or else be stenotic. One or both urethras also may be hypospadiac or epispadiac. A wide range of scrotal abnormalities may be present. Because the duplicated cloacal membrane is likely to be a widened structure, the “wedge” effect could result in the stigmata of covered exstrophy. In some patients, the abnormalities suggest a form of partial caudal duplication with extensive midline defects or duplication involving the derivatives of the allantois, hindgut, and neural tube [24].
It is thought that diphallia occurs in the fetus between the 23rd and 25th days of gestation when an injury, chemical stress, or malfunctioning homeobox genes hamper proper function of the caudal cell mass of the fetal mesoderm as the urogenital sinus separates from the genital tubercle and rectum to form the penis.
Classification
Two distinct forms of penile duplication exist:
The most common form is associated with bladder exstrophy complex. The patient exhibits a bifid penis, which consists of two separated corpora cavernosa that are associated with two independent hemiglands.
The second form, or true diphallia, is an extremely rare congenital condition. It presents in many ways, ranging from duplication of the glans alone to duplication of the entire lower genitourinary tract.
The urethral opening can be in normal position or in a hypospadiac or epispadiac position. Associated anomalies of the GI, genitourinary, and musculoskeletal systems are expected. Because these anomalies are the principal causes of mortality, examining and treating patients for these conditions as soon as possible is important.
According to Schneider [24], diphallia can be divided into four categories:
1.
Duplication of the glans alone
2.
Bifid diphallia
3.
Complete diphallia with each penis having two corpora cavernosa and a corpus spongiosum
4.
Pseudodiphallia in which there is a rudimentary atrophic penis existing independently of the normal penis
Treatment
Treatment must be individualized with consideration of the associated anomalies with the goal of attaining a satisfactory functional and cosmetic result.
2.6 Penile Deviation
Nomenclature
Penile torsion, penile diversion, penile curvature
Definition
Penile torsion is a rotational defect of the penile shaft. Almost always the shaft is rotated in a counterclockwise direction (i.e., to the left side) (Fig. 2.16); right-sided rotation is a rare entity (Fig. 2.17). In most cases, penile size is normal and the condition is unrecognized until circumcision is performed or until the foreskin is retracted. Penile torsion may also be associated with hypospadias or a dorsal hood deformity without a urethral abnormality. In most cases of penile torsion, the median raphe spirals obliquely around the shaft (Fig. 2.18).