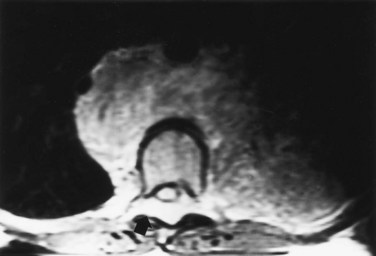
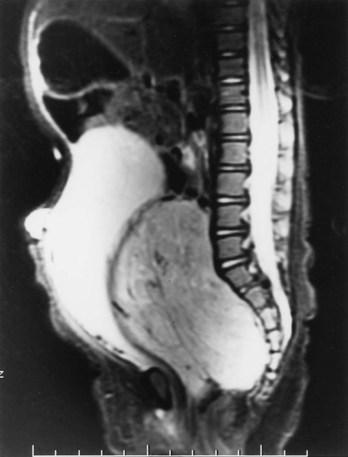
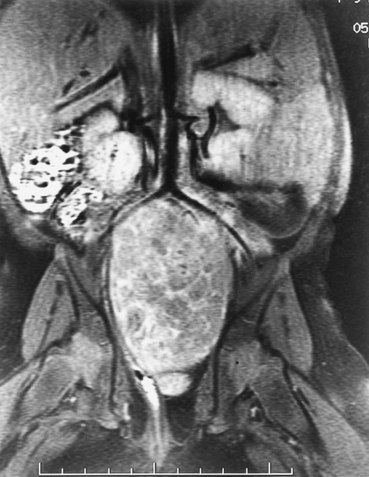
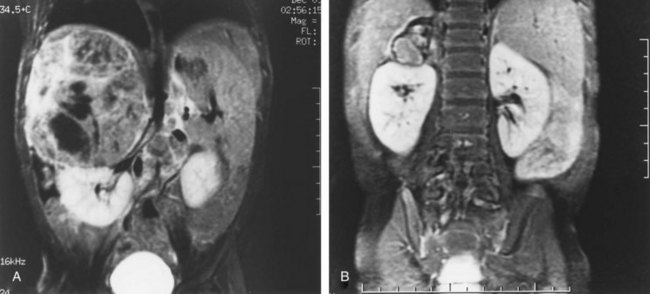
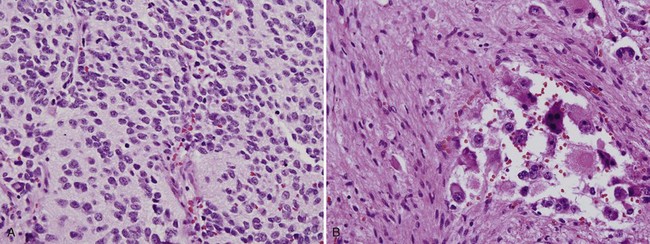
Michael L. Ritchey, MD, FAAP, FACS, Robert C. Shamberger, MD Neuroblastoma is the most common extracranial solid tumor of childhood. Regrettably over half of the children present with metastatic disease. Neuroblastoma is known to arise from cells of the neural crest that form the adrenal medulla and sympathetic ganglia. Tumors may occur anywhere along the sympathetic chain within the neck, thorax, retroperitoneum, or pelvis, or in the adrenal gland. Seventy-five percent arise in the retroperitoneum, 50% in the adrenal, and 25% in the paravertebral ganglia. The variety of locations where these tumors arise and the spectrum of their differentiation results in a wide range of clinical presentations and behaviors (Brodeur, 1991). These tumors can undergo spontaneous regression (Brodeur, 1991), differentiate to benign neoplasms, or exhibit extremely malignant behavior. Neuroblastoma accounts for 8% to 10% of all childhood cancers. In the United States, the annual incidence is 10 cases per 1 million live births. It is the most common malignant tumor of infancy. A recent review of 3666 children enrolled in the cooperative group trials of the Pediatric Oncology Group and the Children’s Cancer Group showed a median age at diagnosis of 19 months (Brodeur and Maris, 2006). Thirty-six percent were infants, 89% were younger than 5 years, and 98% were diagnosed by 10 years of age. There have been a number of familial cases reported, which are postulated to represent an autosomal dominant pattern of inheritance (Knudson and Strong, 1972a; Robertson et al, 1991). Although the median age at diagnosis of neuroblastoma is 19 months, in familial cases it is 9 months (Kushner et al, 1986). At least 20% of patients with familial neuroblastoma have bilateral adrenal or multifocal primary tumors, which are quite unusual in spontaneous cases. The risk for development of neuroblastoma in a sibling or offspring of a patient with neuroblastoma is less than 6% (Kushner et al, 1986). Linkage analysis in seven families with two or more first-degree relatives affected with neuroblastoma identified a single interval at chromosome 16p12-13 with consistent linkage (Maris et al, 2002). This suggests that a hereditary neuroblastoma predisposition gene may be located at this site and may explain the familial cases. Numerous karyotypic abnormalities have been found in neuroblastoma, and these are recognized to have prognostic significance. These changes occur in the form of chromosomal deletions, translocations, and cytogenetic evidence of gene amplification. Aneuploidy of the tumor DNA occurs in a significant number of cases and is a favorable prognostic indicator, whereas amplification of the N-MYC oncogene seen in roughly 20% of primary tumors is an adverse prognostic indicator (Look et al, 1991; Muraji et al, 1993). These findings have been so striking that neuroblastoma was the first tumor in which the intensity of chemotherapy for a patient was determined not only by the stage and histology of the tumor but by its “biologic markers,” which were primarily chromosomal (Matthay et al, 1998). Deletion of the short arm of chromosome 1 (1p) is found in 25% to 35% of neuroblastomas and is an adverse prognostic marker (Brodeur et al, 1992; Caron et al, 1996). The deletions are of various lengths, but, in a series of eight cases, a consensus deletion included the segment 1p36.1-2, suggesting that genetic information related to neuroblastoma tumorigenesis is located in that segment, although the involved genes have not been identified despite intensive investigation (Weith et al, 1989; Maris et al 2007). There have been reports of constitutional abnormalities involving the short arm of chromosome 1 (Laureys et al, 1990). Recently, it has been demonstrated that loss of heterozygosity (LOH) at 1p36 and Unb11q is independently associated with a worse outcome in patients with neuroblastoma (Attiyeh et al, 2005). (Unbalanced LOH implies LOH at markers on 11q with retention of 11p material, in contrast with “whole-chromosome” 11 LOH, where there is LOH at every marker along the chromosome.) Of note, while the 1p deletions are seen in conjunction with advanced stage and N-MYC amplification, the 11q deletions are rarely seen in tumors with N-MYC amplification, but are associated with other high-risk features (Spitz et al, 2006). In future trials of the Children’s Oncology Group (COG), treatment duration will be established by tumor 1p and 11q allelic status. An additional genetic abnormality, gain of one to three copies of 17q, often the result of translocation with chromosomes 1 or 11, has been demonstrated to correlate with more aggressive tumors (Bown et al, 1999). The break points vary, but the addition of a region from 17q22-qter is common suggesting that genes replicated in this region provide an advantage (Schleiermacher et al, 2004). In multivariate analysis, gain of 17q was the most powerful prognostic factor, followed by the presence of stage 4 disease and deletion of 1p. In 1963, Beckwith and Perrin coined the term in-situ neuroblastoma for small nodules of neuroblasts found incidentally within the adrenal gland that are histologically indistinguishable from neuroblastoma (Beckwith and Perrin, 1963). In-situ neuroblastoma was found during postmortem examination in 1 of 224 infants younger than 3 months. This represents an incidence of approximately 40 to 45 times greater than that of clinical tumors, suggesting that these small tumors regress spontaneously in most cases. Studies have shown subsequently that these neuroblastic nodules are found in all fetuses studied and generally regress (Ikeda et al, 1981). Neuroblastoma identified by prenatal ultrasonography (US) has also been shown to have a clinically favorable course (Ho et al, 1993). The concept of in-situ neuroblastoma has been used to support the argument that many neuroblastomas arise and regress spontaneously. This concept has been further supported by population-based studies in Quebec province and in Japan, where prospective screening of infants for neuroblastoma has been performed based on urinary catecholamine excretion. An increased number of children were identified with low-stage neuroblastoma, a higher frequency than present clinically, but there was no decrease in the incidence of advanced-stage tumors seen at an older age (Hayashi et al, 1995; Woods et al, 1996). Evaluation of adrenal tumors resected in the neonatal period, whether cystic or solid, showed that in most the “biologic markers” were favorable (Kozakewich et al, 1998). The highly favorable outcome of infants diagnosed with neuroblastoma in the population screening studies first led to attempts at expectant observation. These trials demonstrated a high rate of spontaneous resolution (Yamamoto et al, 1998; Yoneda et al, 2001). Spontaneous regression of these perinatally identified lesions has also been demonstrated radiographically (Holgerson et al, 1996). A current prospective study by the Children’s Oncology Group is evaluating infants with adrenal masses identified in the perinatal or neonatal period. Expectant observation for infants with small lesions with favorable catecholamine ratios is encouraged. These lesions are then followed closely by serial ultrasound evaluations. The goal of this protocol is to document that expectant observation in these small stage I lesions in infants is safe and appropriate, as has been suggested by smaller retrospective studies. Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma display a histologic spectrum of maturation and differentiation (Fig. 137–1). A grading classification of neuroblastoma, introduced in 1984 by Shimada and subsequently modified by him and others as the International Neuroblastoma Pathology Classification in 1999, has helped to define risk-based subtypes of ganglioneuroblastoma and neuroblastoma (Shimada et al, 1984, 1999a, 1999b). This revised system has been demonstrated to add independent prognostic information beyond the contribution of age that is contained in the system (Sano et al, 2006). Ganglioneuroma is a histologically benign, fully differentiated counterpart of neuroblastoma. It is unclear whether ganglioneuroma arises de novo or by maturation of a preexisting neuroblastoma or ganglioneuroblastoma. Metastatic lesions from neuroblastoma have been observed to develop the histology of mature ganglioneuroma, supporting the latter theory (Hayes et al, 1989). (Courtesy of Dr. Harry Kozakewich.) The Shimada classification is an age-linked histopathologic classification. One of its important aspects is determining whether the tumor is stroma poor or stroma rich. Patients with stroma-poor tumors with unfavorable histopathologic features have a very poor prognosis (less than 10% survival) (Shimada et al, 1984). Stroma-rich tumors can be separated into three subgroups: nodular, intermixed, and well differentiated. Tumors in the latter two categories more closely resemble ganglioneuroblastoma or immature ganglioneuroma and carry a higher rate of survival. The stroma-poor tumors can be divided into favorable and unfavorable subgroups based on the patient’s age at diagnosis, the degree of histologic maturation, and the mitotic rate. When compared with other clinical features, these histologic patterns were independently predictive of outcome (Shimada et al, 1984). In contrast to neuroblastomas, ganglioneuromas are most often diagnosed in older children and are usually located in the posterior mediastinum and retroperitoneum, with only a small number arising in the adrenal glands (Enzinger and Weiss, 1988). Ganglioneuromas often grow to a very large size before they cause symptoms as a result of compression of adjacent structures or extension into the spinal canal (Benjamin et al, 1972) (Fig. 137–2). Most primary tumors arise within the abdomen (65%); the frequency of adrenal tumors is slightly higher in children than in infants. Physical examination often reveals a fixed, hard abdominal mass. Pelvic neuroblastoma arising from the organ of Zuckerkandl accounts for 4% of tumors (Haase et al, 1995). Extrinsic compression of the bowel and bladder can produce symptoms of urinary retention and constipation (Fig. 137–3). Metastases are present in 70% of patients with neuroblastoma at diagnosis and can be responsible for a variety of the clinical signs and symptoms at presentation. A number of unique paraneoplastic syndromes have been associated with both localized and disseminated neuroblastoma. Symptoms produced by catecholamine release may mimic those seen in pheochromocytoma: paroxysmal hypertension, palpitations, flushing, and headache. Secretion of vasoactive intestinal peptide (VIP) by the tumor can produce severe watery diarrhea and hypokalemia (Cooney et al, 1982). Another unusual presentation of neuroblastoma is acute myoclonic encephalopathy, in which patients develop myoclonus, rapid multidirectional eye movements (opsoclonus), and ataxia. It is thought to result from an interaction of antibodies produced against the neuroblastoma to normal neural tissues (Farrelly et al, 1984; Connolly et al, 1997). Although this syndrome is associated with a favorable outcome from an oncologic perspective (Altman and Baehner, 1976), prolonged neurologic impairment, including learning disabilities and developmental delay, is the rule in 70% to 80% of these children, and symptomatic therapy is often required (Russo et al, 1997; Rudnick et al 2001; Mitchel et al 2002). Adrenocorticotropic hormone (ACTH) or steroids are the basis of most therapy, but other treatments include high-dose intravenous gammaglobulin and cyclophosphamide. A current protocol of the Children’s Oncology Group is evaluating the efficacy of intravenous gamma globulin and corticosteroids in the treatment of these children. When sensitive techniques are used, increased levels of urinary metabolites of catecholamines, vanillylmandelic acid (VMA) and homovanillic acid (HVA), are found in 90% to 95% of patients (Williams and Greer, 1963). Therapy with various modalities produces a reduction in catecholamine metabolite excretion in most patients (Gerson and Koop, 1974). These metabolites can be monitored to detect tumor relapse and response to therapy. Anemia is noted in children with widespread bone marrow involvement. Studies suggest that marrow biopsies add substantially to the detection of marrow involvement by tumor, compared with marrow aspirates alone (Franklin and Pritchard, 1983). It is recommended that two marrow aspirates and two biopsies be performed. In the future, neuroblastoma-specific immunocytology of marrow aspirates may obviate the need for marrow biopsies in most patients (Hsiao et al, 1990). Imaging studies play an important role in the evaluation of a child with neuroblastoma. Plain radiographs may demonstrate a calcified abdominal or posterior mediastinal mass. Computed tomography (CT) and magnetic resonance imaging (MRI) provide more information about the local extent of the primary tumors. Invasion of the renal parenchyma is not common, but it can be detected radiographically by CT (Albregts et al, 1994). MRI has advantages over CT in the evaluation of intraspinal tumor extension (see Fig. 137–2) and in demonstrating the relationship between the major vessels and the tumor (Azizkhan and Haase, 1993) (Fig. 137–4). The finding of intratumoral calcifications, vascular encasement, or both on preoperative computed tomography may help distinguish neuroblastoma from Wilms tumor (Dickson et al, 2008). Current Children’s Oncology Group protocols require both a radionuclide bone scan and meta-iodobenzylguanidine (MIBG) scans for staging, but no longer require the classic skeletal survey. MIBG scans use 131I-MIBG (Geatti et al, 1985), which is taken up by the adrenergic secretory vesicles of the tumor cells in both primary and metastatic sites. MIBG scintigraphy can determine the extent of disease and detect tumor recurrence after completion of therapy (Geatti et al, 1985). Recently, 131I-MIBG has been shown to be more sensitive than either 131I-MIBG or bone scans in detecting tumor relapse (Kushner et al, 2009). Osseous metastatic lesions occur most commonly in the long bones and skull. Mass population screening for neuroblastoma has been widely used in Japan for more than 20 years (Nishi et al, 1987). The goal of screening programs is to detect disease at an earlier stage and to decrease the number of older children with advanced-stage disease, thereby improving survival. In fact, the children diagnosed as a result of screening studies have had almost uniformly favorable survival (>97%) (Suita, 2002). An increased number of infants younger than 1 year of age have been diagnosed through the mass screening program (Ishimoto et al, 1990), and most of these patients have lower-stage tumors (Sawada, 1992). Before mass screening started, 20% of neuroblastoma cases were diagnosed before 1 year of age, compared with 55% after its implementation. However, the number of children older than 1 year of age diagnosed with advanced-stage disease has not decreased. These results suggested that the aggressive advanced-stage tumors in older children did not arise from the low-risk tumors seen in the infants less than 1 year of age. There are biologic differences between tumors diagnosed by screening and those detected clinically (Hayashi et al, 1992). In one review of 48 cases discovered by screening, no tumors were observed to have amplified N-MYC oncogene expression (Ishimoto et al, 1991), and in a second review of 20 infants, only one who did poorly had amplification (Hase et al, 2002). Furthermore, 80% had a diploid chromosome pattern, which is also associated with a favorable prognosis. On follow-up, all 48 patients were still alive without tumor. In another series of 357 patients whose tumors were diagnosed by mass screening, the overall survival rate was 97% (Sawada, 1992). Given the favorable biologic characteristics of tumors discovered by screening, it is possible that many would spontaneously resolve without therapy, particularly given the increased incidence of neuroblastomas seen in the screened population. Two large prospective population based studies were conducted in the Province of Quebec and in Germany. These studies demonstrated that urine screening at various ages was successful in identification of neuroblastoma, but there was no decrease in the occurrence of neuroblastoma in older children and its subsequent mortality (Schilling et al, 2002; Woods et al, 2002). Staging of neuroblastoma is an important aspect of management. The stage of the disease is a significant prognostic variable that determines adjuvant therapy. The International Neuroblastoma Staging System (INSS) is based on clinical, radiographic, and surgical evaluation of children with neuroblastoma (Brodeur et al, 1993) (Table 137–1). Earlier staging systems provided generally comparable results in terms of distinguishing low-stage, good-prognosis disease from high-stage, poor-prognosis disease. Use of a uniform international system, however, makes comparison of results from various studies much easier to compare. The biggest differences arise when the various systems are applied to those with intermediate-stage disease. It is in this cohort of children where use of the Risk Group Classification, which combines pathologic findings, stage, and several of the biologic markers, best defines the child’s risk for progressive disease (Table 137–2) (Katzenstein and Cohn, 1998). Although the classification appears complex, it provides the most accurate assessment of how intense the chemotherapy and radiotherapy must be to cure the child. Table 137–1 International Neuroblastoma Staging System * The midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column. A recent effort has been made to identify, preoperatively, those tumors that are resectable without significant surgical risks. The Localized Neuroblastoma European Study Group (LNESG) defined the radiographic criteria associated with higher surgical morbidity. They validated that their presence was associated with a lower rate of complete resection and a greater risk of surgical complications (Cecchetto et al, 2005). This finding has been confirmed by others (Simon et al, 2008). Age remains an important indicator of outcome, as originally reported by Breslow (Breslow and McCann, 1971). Children age 1 year or younger have a better survival than older children (Nitschke et al, 1988). This may be attributed to more favorable biologic parameters in tumors diagnosed at this age. Recent retrospective reviews have suggested that age is a truly more of a “continuous” prognostic variable and that a cutoff of 460 days maximized the outcome difference between the younger and older patients (London et al, 2005). Teenagers and adults with neuroblastoma have a particularly indolent and relentless disease (Gaspar et al, 2003; Kushner et al, 2003). The site of origin is of significance, with better survival noted for nonadrenal primary tumors (Haase et al, 1995). Most children with thoracic neuroblastoma present at a younger age with localized disease and have improved survival even when corrected for age and stage (Adams et al, 1993). Tumors at this site are less likely to have N-MYC amplification and more likely to have a DNA index greater than 1.0, both favorable prognostic indicators (Morris et al, 1995). Stage of the disease is a powerful independent prognostic indicator. Virtually all stage I patients with complete resection of the primary tumor survive. Stage II patients also have a favorable survival prospect, even though there may be incomplete excision (Matthay et al, 1989). Children with advanced regional disease, stages III or I, fare less well and require more intensive treatment. The proportion of patients presenting with localized, regional, or metastatic disease is age dependent (Nitschke et al, 1988). The overall prognosis for stages I, II, or IV-S has a survival range of 75% and 90%, whereas those children with stage IV disease have a 2-year disease-free survival range of 19% to 30% despite intensive therapy, including bone marrow transplantation. The outcome for infants younger than 1 year of age is substantially better than for older patients with the same stage of disease. Stage IV-S (S meaning special) is a distinct category referring to infants with small primary tumors and liver, skin, and bone marrow metastases without radiographic evidence of bone metastases. It was first proposed by Evans and colleagues in 1971. This group of patients has a good prognosis, with overall survival ranging from 80% to 88%. The INSS criteria later restricted this stage to children with less than 10% bone marrow involvement (Brodeur et al, 1993). This stage accounts for 7% to 10% of all cases of neuroblastoma. Many of these tumors undergo spontaneous regression (Evans et al 1987; Haas et al, 1990). In general, the tumors in children with stage IV-S neuroblastoma have favorable prognostic findings not typically seen in children with stage IV disease (Hachitanda et al, 1991; Nickerson et al, 2000). Poor outcome in stage IV-S patients is associated with elevated serum neuron-specific enolase (>100 nmol/mL), ferritin (>280 ng/mL), urinary dopamine levels (>2500 nmol/mmol creatinine), as well as N-MYC amplication and chromosome 1p deletion (Schleiermacher et al, 2003). Most deaths in this group are in infants under 2 months of age with extensive abdominal involvement and with respiratory compromise or disseminated intravascular coagulation (Nickerson et al, 2000). The presence of homogeneously staining regions and double-minute chromosomes was noted in approximately one third of neuroblastoma tumors. These abnormalities are cytogenic manifestations of gene amplification, and it was subsequently found that the N-MYC oncogene was mapped to these regions. The association of N-MYC amplification with the pathogenesis of neuroblastoma is unclear, but N-MYC amplification is almost always present at the time of diagnosis (Brodeur, 1991). Seeger and colleagues (1985, 1988) showed that N-MYC amplification is associated with rapid tumor progression and a poor prognosis. Amplification is found in 5% to 10% of patients with low-stage or stage IV-S (Hachitanda et al, 1991) but in 30% to 40% of those with advanced-stage disease (Brodeur and Fong, 1989; Brodeur, 1990). The poor prognosis associated with N-MYC amplification is independent of patient age or stage of disease at presentation (Iehara et al, 2006). However, not all patients with a poor outcome have N-MYC amplification. Many advanced-stage tumors lack N-MYC at diagnosis, and recurrence or progression of disease develops in most of these patients. DNA content of tumor cells and ploidy number has been reported to have prognostic value in patients with neuroblastoma (Cohn et al, 1990). Studies of DNA content measured by flow cytometry showed that a “hyperdiploid” karyotype (or increased DNA content) was associated with a favorable outcome (Look et al, 1984; Kusafuka et al, 1994). DNA diploidy and tetraploidy were associated with decreased survival. A recent review of 2660 children with localized neuroblastoma registered with the International Neuroblastoma Risk Group database evaluated tumor ploidy in children with N-MYC–amplified tumors (Bagatell et al, 2009). They reported an improved prognosis in children with hyperdiploid tumors compared to diploid tumors. This could be used to identify children for reduction in therapy. Deletions of the short arm of chromosome 1 have been found in 70% to 80% of the near-diploid tumors that have been karyotyped (Brodeur and Fong, 1989; Brodeur, 1990). Preliminary studies suggest a correlation between 1p deletion and poor survival (Brodeur and Fong, 1989; Hayashi et al, 1989). Because there is an association between N-MYC amplification and 1p deletion, it remains to be determined whether this finding has independent prognostic significance. Children currently treated on protocols of the COG are assigned to a risk group that is determined by age, stage of disease, N-MYC status, histologic grade, and DNA ploidy (Katzenstein et al, 1998) (see Table 137–2). Other factors that have been demonstrated to have prognostic significance, although they are often associated with these genetic abnormalities, include expression of the gene encoding the high-affinity nerve growth factor receptor (termed TRKA proto-oncogene) and the low-affinity nerve growth factor receptor (Tanaka et al, 1995). Both are favorable prognostic predictors and are inversely related to amplification of N-MYC oncogene (Nakagawara et al, 1993). Lack of expression of CD44 glycoprotein on the tumor cell surface and increased levels of serum ferritin, serum neuron-specific enolase, and serum lactate dehydrogenase are all adverse prognostic factors (Chan et al, 1991; Silber et al, 1991). They have not, however, been shown by multivariate analysis to be independently predictive from age, stage, ploidy, and N-MYC status. Telomere length has also been described as a significant prognostic parameter, and in a cohort of high-risk patients, it was the sole significant parameter that identified a group of patients with a favorable prognosis (Ohali et al, 2006). The treatment modalities primarily used in the management of neuroblastoma are surgery, chemotherapy, and radiation therapy. The role of each in individual patients varies depending on tumor stage, age, and biologic prognostic factors. These can be used to stratify patients into favorable and unfavorable categories by risk group (see Table 137–2). Children with stage I neuroblastoma have a disease-free survival rate of greater than 90% with surgical excision alone (O’Neill et al, 1985; Nitschke et al, 1988; DeBernardi et al, 1995). Chemotherapy is indicated only in the event of recurrence unless the child has N-MYC amplification and unfavorable histology. The Pediatric Oncology Group reviewed 101 children with localized neuroblastoma who had complete gross excision of the primary tumor (Nitschke et al, 1988). Nine patients experienced relapse, but six were salvaged with chemotherapy. Radiation therapy has no role in this subset of patients. With current use of the risk factor grading, those children with recurrence in the past may be identified now as the small number with adverse biologic markers. In a comparable study from the Children’s Cancer Group, 374 stage I and II patients were treated primarily by resection (Perez et al, 2000). Event-free survival (EFS) and overall survival (OS) for stage I patients were 93% ± 3% and 99% ± 1%, respectively, compared with 81% ± 4% and 98% ± 2% for stage II patients, respectively. Supplemental treatment was required in only 10% of stage I and 20% of stage II patients, and excellent overall survival was achieved in the stage II patients. N-MYC amplification, unfavorable histology, age greater than 2 years, and positive lymph nodes predicted a lower overall survival. Radical resection resulting in removal of normal organs, particularly the kidney, is not justified in this group of patients. Radiation of the local tumor bed has been advocated for treatment of residual disease in stage II cases. However, a review of 156 patients with stage II neuroblastoma found a 90%, 6-year, progression-free survival rate regardless of whether radiation therapy was used (Matthay et al, 1989). Therefore radiation should be reserved for those patients who fail to respond to either primary or secondary chemotherapy. In stage III disease, or in stage II with extensive tumor around the kidney and renal vessels, preoperative treatment with chemotherapy significantly decreases the risk of nephrectomy as a result of resection of the tumor (Shamberger et al, 1998) (Fig. 137–5). The generally favorable behavior of IV-S disease has been explained with the understanding of biologic markers. The vast majority of these infants have tumors with entirely favorable markers, explaining their favorable behavior. However, a small percentage of these patients have adverse markers, and it is these children who have progressive disease that often is fatal. Resection of the primary is not mandatory (Nickerson et al, 1985; Evans et al, 1987). Although excellent survival has been reported after surgery (Martinez et al, 1992), information regarding histologic prognostic factors was not available for all of these patients. In a review of 110 infants with stage IV-S disease, the entire cohort had an estimated 3-year survival rate of 85% ± 4% (Katzenstein et al, 1998). This rate was significantly decreased to 68% ± 12% for infants whose tumors were diploid; to 44% ± 33% for those with N-MYC amplification; and to 33% ± 19% for those with unfavorable histology. Of note, there was no statistical difference in survival rate for infants who underwent complete resection of their primary tumor compared with those who had partial resection or only biopsy (Katzenstein et al, 1998; Nickerson et al, 2000; von Schweinitz et al, 2002). Patients with extensive metastatic disease and N-MYC amplification represent a high-risk group (Martinez et al, 1992). These patients should be considered for a more aggressive treatment with multimodal therapy, according to the risk group classification (see Table 137–2) (Schleiermacher et al, 2003). Those with favorable biologic markers and no symptoms can be followed with supportive care and limited chemotherapy, and hepatic radiation to infants with significant symptoms. Intensive chemotherapy is reserved for those with adverse markers, although these infants do poorly even with therapy. There is debate regarding the extent of surgical resection that is required for stage III lesions. A report from the Children’s Cancer Group of 58 patients with stage III disease found that 8 of 12 patients with initial complete excision, and 12 of 14 with subsequent resection of the primary tumor, were long-term survivors (Haase et al, 1989). This result contrasts with only 9 of 32 survivors among patients in whom complete tumor excision could not be accomplished. Significant morbidity was reported in association with the surgical procedures, including 21 major complications. The Italian Cooperative Group for Neuroblastoma found that complete resection after chemotherapy of extensive unresectable neuroblastoma was associated with improved survival, compared with partial resection only (Garaventa et al, 1993). Similar results have been noted by others (LeTourneau et al, 1985; O’Neill et al, 1985; Powis et al, 1996). It has been suggested by some that even children with stage III disease do not need cytotoxic therapy if the biologic marker N-MYC amplification is not present (Kushner et al, 1996). These results are not widely accepted, however, and confirmatory studies are required before this policy can be widely adopted. Children with bulky pelvic tumors generally do quite well even with limited residual disease (LeClair et al, 2004). Extensive surgery at this site has been associated with long-term neurologic sequelae; thus the extent of resection must be balanced against this morbidity (Cruccetti et al, 2000). There is conflicting evidence for the role of extensive resection in stage IV disease between studies that support (LeTourneau et al, 1985; Haase et al, 1991; Tsuchida et al, 1992; LaQuaglia et al, 1994; Chamberlain et al, 1995; DeCou et al, 1995; Yokoyama et al, 1995; Kuroda et al, 2003; Adkins et al, 2004; LaQuaglia et al, 2004; Koh et al, 2005; McGregor et al, 2005) and those that refute that approach (Sitarz et al, 1983; Matsumura et al, 1988; Adams et al, 1993; Losty et al, 1993; Kiely, 1994; Kaneko et al, 1997; Castel et al, 2002; von Schweinitz et al, 2002). In a retrospective review, Kiely (1994) compared the results of radical tumor resection with those of more conventional surgery in patients with stage III and IV disease. Kiely found no difference in survival between 46 patients treated with radical surgical procedures and 34 patients treated with more conventional surgery. Shorter and colleagues (1995) also did not find any evidence that the extent of surgical resection had an impact on the survival of stage IV patients. In these nonrandomized studies, it has been difficult to determine whether improved survival in those with complete resection has been due to the more favorable intrinsic biology of the tumor allowing resection or truly due to the completeness of resection. As the intensity of the therapy increases, including the use of autologous bone marrow transplantation and control of distant metastasis, the impact of maximal local control may become apparent. The combination of gross total resection and external beam irradiation has achieved local control in 84% to 90% of children (Wolden et al, 2000; Kushner et al, 2001). In another series of reports, intensive chemotherapy followed by double autologous bone marrow transplantation, aggressive surgical resection, and radiotherapy has achieved an overall survival of 56% and a local recurrence rate of only 2.6% (Marcus et al, 2003; von Allmen et al, 2005). Usually the safest approach for advanced tumors is to defer resection until after initial chemotherapy (Berthold et al, 1989; Shamberger et al, 1991; Shochat, 1992; LaQuaglia et al, 1994; Black et al, 1996). The tumors are smaller and firmer, with less risk of rupture and hemorrhage following chemotherapy, resulting in a decreased rate of complications, particularly nephrectomy (Shamberger et al, 1998) (see Fig. 137–5). One specific complication that is encountered after resection of extensive tumor surrounding the celiac axis and the superior mesenteric artery is diarrhea (Kiely, 1994). It is thought to result from resection of the autonomic nerves to the gut found anterior to the aorta at the base of the superior mesenteric artery and the celiac axis (Rees et al, 1998). Preoperative chemotherapy does appear to increase the proportion of children able to achieve a complete resection (Adkins et al, 2004). Surgery usually is performed 13 to 18 weeks after initiation of chemotherapy, allowing three to four courses of treatment (Azizkhan and Haase, 1993). Some tumors remain inoperable even after chemotherapy. In a large cohort (134 infants), N-MYC amplification, serum ferritin, Shimada histopathologic classification, and bone marrow involvement by immunocytology were analyzed. Although each factor had prognostic significance by univariate analysis, only N-MYC was significant by multivariate analysis; the EFS for infants without N-MYC amplification was 93% ± 4% versus 10% ± 7% in those with amplification despite intensive therapy (Schmidt et al, 2000). Recent trials in Europe evaluated these infants without N-MYC amplification and achieved excellent survival (OS of 97.6% at 2 years) without chemotherapy in those with primaries extending across the midline or a positive skeletal survey (DeBernardi et al, 2009). Infants with overt metastases to the skeleton, lung, and central nervous system (CNS) were treated with a minimum of four chemotherapy courses and achieved a 2-year OS of 95.6%. A variety of multiagent treatment regimens have been developed to treat high-risk patients with neuroblastoma. The goal of this treatment intensification is better disease control. Although initial response rates are improving, with a prolonged time to progression of disease, relapse continues to be a major problem, and the 4-year overall survival rate in stage IV disease is 20% (Ikeda et al, 1989; Haase et al, 1991). The dose intensification of chemotherapy needed for local tumor control results in significant myelosuppression, limiting the amount of therapy that can be given. This has prompted the use of autologous bone marrow transplantation after sublethal chemotherapy or total-body irradiation. The use of marrow-ablative chemoradiotherapy followed by autologous marrow reinfusion has resulted in complete remission in up to 50% of patients with recurrent stage IV disease (Moss et al, 1987; Seeger et al, 1991; Dinndorf et al, 1992; Mugishima et al, 1994; Matthay et al, 1995; Grupp et al, 2000). However, a significant problem is the risk for late relapse. The randomized clinical trial of the German Society of Paediatric Oncology and Hematology of high-risk neuroblastoma showed improved event-free survival at 3 years with myeloablative therapy and autologous stem cell rescue compared with maintenance chemotherapy (Berthold et al, 2005). These results were similar to those of the COG study (Matthay et al, 1999). Regrettably, the overall survival rates were not statistically different in either study at initial report. A recent update of the latter study reveals that the 5-year OS of the patients receiving the autologous bone marrow transplant was improved (Matthay et al, 2009). New agents in phase I and II trials for relapsed neuroblastoma include temozolomide, irinotecan, and topotecan (Kushner et al, 2006; Rubie et al, 2006; Simon et al, 2007). As efficacy is established, they will be advanced into clinical trials for high-risk tumors with a goal to improve the overall outcome. Because increasing the intensity of chemotherapy appears to have reached its limit with the use of double autologous bone marrow transplantation, other routes of treatment must be identified. The use of biologic modifiers is being investigated (Villablanca et al, 1995). 13-cis-Retinoic acid produces differentiation of neuroblastoma in cell cultures. It was given for a 6-month period after cytotoxic therapy in children with advanced-stage disease and significantly decreased the frequency of relapse (Matthay et al, 1999). Long-term follow up has shown that the 5-year OS is improved with the use of 13-cis–retinoic acid in these children after transplant or intensive chemotherapy (Matthay et al, 2009). Other avenues of treatment in current phase I and phase II trials include vaccine and antibody therapy against the GD2 cell surface marker occurring in neuroblastoma. A new synthetic retinoid, fenretinide, which has produced apoptosis rather than differentiation in neuroblastoma cell lines, is also in early clinical trials. Another modality in the treatment of metastatic neuroblastoma is the use of 131I-MIBG (Hutchinson et al, 1992). The finding that both the primary tumor and metastatic areas take up this radiotracer suggested the possibility that therapeutic doses can be delivered to the tumor. Preliminary analysis indicates that objective responses do occur in terms of reduction of tumor volume, even in previously heavily treated patients (Howard et al, 2005; Matthay et al, 2007). Significant myelosuppression is seen with dose escalation, however, and stem cell support is required. Radiotherapy is effective for local control in neuroblastoma and risk of local relapse can be correlated with the biologic markers. Although irradiation has not provided a benefit in low-stage tumors, it has increased local control in children with advanced stage IV or bulky stage III tumors (Matthay et al, 1989; Castleberry 1991; Evans et al, 1996). A randomized control trial to evaluate the efficacy of local control between radiotherapy alone and surgery has not been performed. Doses of external beam irradiation used have ranged between 15 and 30 Gy, depending on the patient’s age, location, and extent of residual disease. Intraoperative radiation therapy has been used for patients with unresectable disease. This technique has the advantage of delivering a higher dose of radiation to the operative field while sparing normal adjacent tissues (Leavey et al, 1997). Although its use has been promoted, it has not been convincingly demonstrated to improve control when compared with external beam irradiation (Haas-Kogan et al, 2000). Extension of tumor into the spinal canal produces symptoms of spinal cord compression in up to 5% of patients presenting with neuroblastoma (DeBernardi et al, 2001), while up to 13% of patients have radiographic evidence of extension into the spinal canal (Plantaz et al, 1996). These children have been treated by decompressive laminectomy, radiotherapy, or chemotherapy. Neurologic outcome has been similar by all modalities and, regrettably, patients presenting with severe motor deficits generally recover little function (DeBernardi et al, 2001; Katzenstein et al, 2001). Because of the delayed complications of scoliosis after laminectomy, current recommendations are to initiate treatment with chemotherapy and reserve laminectomy for children with progressive neurologic deterioration (Katzenstein et al, 2001). Radiotherapy is now generally avoided, because of its adverse effect on growth of the spine. Key Points Neuroblastoma Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in infants and children, with nearly 350 new cases in the United States each year. It is the third most common solid tumor in children after neuroblastoma and Wilms tumor. Fifteen to 20 percent of all RMS arise from the genitourinary system (Maurer et al, 1988; Crist et al, 2001). The most common genitourinary sites are the prostate, bladder, and paratestisticular; the vagina and uterus are relatively unusual sites. Survival rates vary with site; certain sites such as the vagina and paratesticular have a better prognosis than bladder/prostate primaries (Rodary et al, 1988; Crist et al, 1990; Martelli et al, 1999; Arndt et al, 2001; Wiener et al, 2001; Ferrari et al, 2002; Meza et al, 2006). There is a bimodal age distribution with a peak incidence in the first 2 years of life and again at adolescence, but two thirds of cases occur in children under 6 years of age (LaQuaglia et al, 1994). The botryoid variants arising from the bladder and vagina are seen almost exclusively in young children. Subgroups of children with a genetic predisposition to the development of RMS have been identified. The Li-Fraumeni syndrome associates childhood sarcomas with mothers who have an excess of premenopausal breast cancer and with siblings who have an increased risk of cancer (Li and Fraumeni, 1969). A mutation of the TP53 tumor suppressor gene was found in the tumors in all patients with this syndrome (Malkin et al, 1990). An increased incidence of RMS has been found in association with neurofibromatosis (Sung et al, 2004). The two major histologic subtypes of RMS, embryonal and alveolar, have been noted to have distinct cytogenetic abnormalities. Alveolar RMS is associated with a translocation between chromosomes 1 or 2 and 13, resulting in the formation of a chimeric protein (Parham, 1994). PAX3, a DNA-binding protein on chromosome 2, or PAX7, a DNA-binding protein and on chromosome 1, is fused to the FKHR gene on chromosome 13 (Anderson et al, 2001). About 25% of cases lack these translocations. These genes may be involved in the pathogenesis of alveolar rhabdomyosarcoma and are prognostic factors as well. Expression of PAX3-FKHR is an adverse prognostic factor for children presenting with metastatic alveolar RMS (Sorensen et al, 2002). In addition, patients with the t(1;13) translocation, PAX7-FKHR fusion, are often younger and have a better prognosis than do their counterparts with the t(2;13), PAX3-FKHR fusion abnormality (Kelly et al, 1997; Anderson et al, 2001; Sorensen et al, 2002). Embryonal RMS demonstrates loss of heterozygosity (LOH) on chromosome 11p15.5 at a different location than the WT2 gene implicated in the development of some Wilms tumors (Douglass et al, 1987; Scrable et al, 1990). This region is the location of the IGF-2 gene. IGF-2 overexpression has been documented in alveolar and embryonal rhabdomyosarcoma (Leiroth et al, 1995). IGF-2 is a growth factor that can stimulate the growth of RMS tumor cells, and antibodies directed against IGF-2 can inhibit tumor growth (El-Badry et al, 1990). Amplification of the transcription factor N-MYC has been noted in neuroblastoma and in RMS (Williamson et al, 2005). Overexpression of N-MYC was associated with an adverse outcome in alveolar RMS. The Intergroup Rhabdomyosarcoma Study Group (IRSG) has developed a pathology classification recognizing three major histologic groups that have prognostic significance (Asmar et al, 1994). Embryonal RMS is the most common subtype of RMS and accounts for most of the genitourinary tumors (Maurer et al, 1977; Newton et al, 1988). This may occur in solid form arising in muscle groups, such as the trunk and extremities, or as the so-called sarcoma botryoides, a polypoid variety that occurs in hollow organs or body cavities, such as the bladder or vagina. Embryonal RMS may have variable cell types with small, round cells with hyperchromatic nuclei and large, polygonal-shaped cells with abundant eosinophilic cytoplasm (Fig. 137–6). There is a spindle cell, or leiomyomatous, variant seen frequently in the paratesticular region. The botryoid and spindle cell variants of embryonal RMS are associated with an excellent survival. The second most common form is alveolar, comprising 15% to 20% of cases. Alveolar RMS occurs more commonly in the trunk and extremities than in genitourinary sites and has a worse prognosis than embryonal tumors (Hays et al, 1983; Newton et al, 1988; Crist et al, 2001; Stevens et al, 2005). Alveolar RMS also has a higher rate of local recurrence and spread to regional lymph nodes, bone marrow, and distant sites. The third category consists of undifferentiated tumors that also fare poorly. Figure 137–6 Embryonal rhabdomyosarcoma with spindle-shaped cells that have a stromal-rich appearance. The diagnosis of rhabdomyosarcoma can occasionally be difficult with conventional histologic techniques. In such cases, histology may be complemented by other studies, including electron microscopy, cytogenetics, DNA flow cytometry, and immunochemistry (Shapiro et al, 1991; Davicioni et al, 2009). Immunohistochemical staining has been shown to be an effective adjunct for the diagnosis of alveolar RMS (Dias et al, 2000). Genes of the MyoD family are important in the differentiation of skeletal muscle (Parham, 1994). MyoD is important in the switch from proliferation to differentiation, and loss of this control and excess proliferation could lead to RMS (Sebire and Malone, 2003). Fetal myoblasts express MyoD and myogenin, another myogenic regulatory gene, whereas adult myoblasts do not. MyoD and myogenin are expressed in RMS and is believed to represent failure of differentiation. Myogenin has been proven to be a useful marker of alveolar RMS. Alveolar RMS demonstrates high levels of staining, whereas embyronal RMS is either negative or has a low level of staining (Dias et al, 2000). Diffuse myogenin expression by immunohistochemistry has been shown to be predictive of decreased survival independent of tumor stage, site or histology (Heerema-McKenney et al, 2008). Tumor stage at diagnosis is most predictive of clinical outcome, because patients with localized disease have a better prognosis (Lawrence et al, 1987). The likelihood of regional lymph node extension varies with site of the primary tumor, approaching 50% for older boys with paratesticular tumors. The most common site of metastatic spread of RMS is the lungs. The clinical group staging system was developed by the IRSG in 1972 (Table 137–3). One of the difficulties inherent in this system is that the group is dependent to a large extent on the completeness of surgical excision. As the treatment of RMS has evolved, more patients undergo biopsy only at the initial surgical procedure, leaving gross residual disease. This results in the shifting of more patients from group I to group III. Biologically equivalent tumors could end up in different categories, depending on the aggressiveness of the initial surgical resection. Table 137–3 Intergroup Rhabdomyosarcoma Study Group Clinical Grouping Classification A site-specific TNM staging system was devised by the IRSG for IRS-IV (Table 137–4) (Lawrence et al, 1997). The TNM system segregates patients by favorable and unfavorable sites. Favorable GU sites include the paratesticular and the vulvar-vaginal-uterine ones. Unfavorable GU sites are the bladder and prostate. Presence of distant metastases at diagnosis, involved regional lymph nodes, and large primary tumors (>5 cm) at unfavorable sites are relatively unfavorable prognostic signs (Lawrence et al, 1997
Neuroblastoma
Epidemiology and Genetics
Incidence
Genetics
Constitutional Chromosome Abnormalities
Embryology and Spontaneous Regression
Pathology
Clinical Presentation and Pattern of Spread
Diagnosis
Laboratory Evaluation
Imaging
Screening
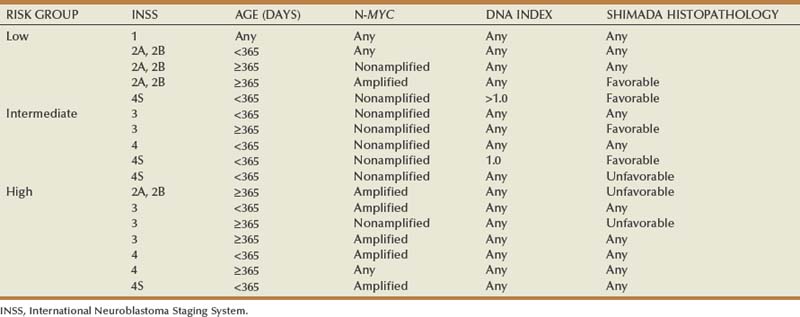
Staging
STAGE
DEFINITION
1
Localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes negative for tumor microscopically (nodes attached to and removed with the primary tumor may be positive).
2A
Localized tumor with incomplete gross excision; representative ipsilateral nonadherent lymph nodes negative for tumor microscopically.
2B
Localized tumor with or without complete gross excision, with ipsilateral nonadherent lymph nodes positive for tumor; enlarged contralateral lymph nodes must be negative microscopically.
3
Unresectable unilateral tumor infiltrating across the midline,* with or without regional lymph node involvement; or localized unilateral tumor with contralateral regional lymph node involvement; or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement.
4
Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin, and/or other organs.
4S
Localized primary tumor (as defined for stage 1, 2A, or 2B), with dissemination limited to skin, liver, and/or bone marrow (less than 10% tumor) in infants less than 1 year of age.
Clinical Variables
Biologic Variables
Treatment
Surgery
Low-Risk Disease (Stages I, II, and IV-S)
High-Risk Disease (Stages III and IV)
Chemotherapy
New Innovative Biologic Therapies
Radiotherapy
Spinal Cord Compression
Genitourinary Rhabdomyosarcoma
Etiology, Epidemiology, and Genetics
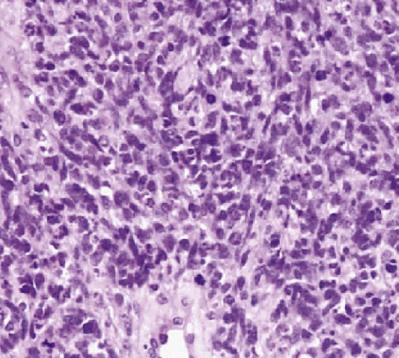
Pathology and Patterns of Spread
Clinical Grouping and Staging
Group I
Localized disease completely resected
Confined to organ of origin
Contiguous involvement
Group II
Total gross resection with evidence of regional spread
Microscopic residual
Positive nodes but no microscopic residual
Positive nodes but microscopic residual in nodes or margins
Group III
Incomplete resection with gross residual disease
After biopsy only
After gross or major resection of the primary (>50%)
Group IV
Distant metastasis at diagnosis (lung, liver, bones, bone marrow, brain, and nonregional nodes)
Positive cytology in cerebrospinal fluid, pleural or peritoneal fluid, or implants on pleural or peritoneal surfaces are regarded as stage IV
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pediatric Urologic Oncology