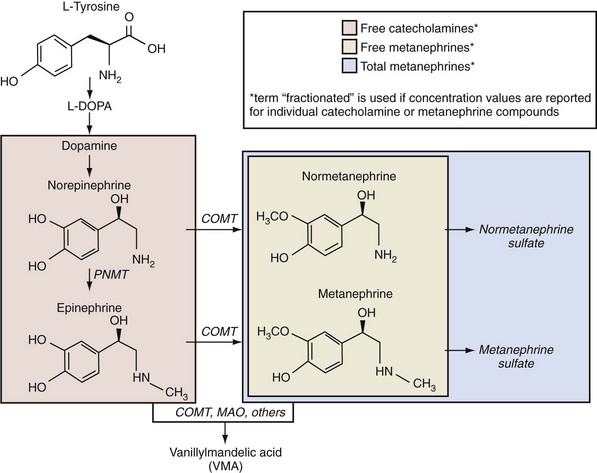
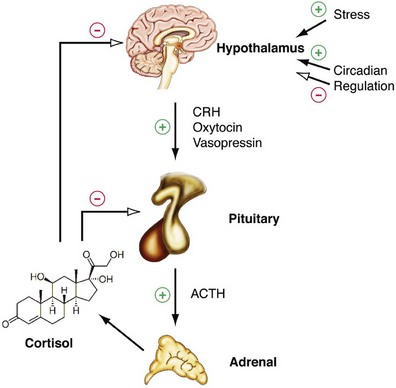
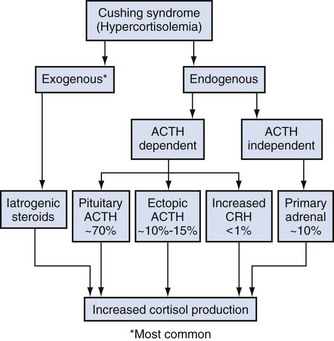
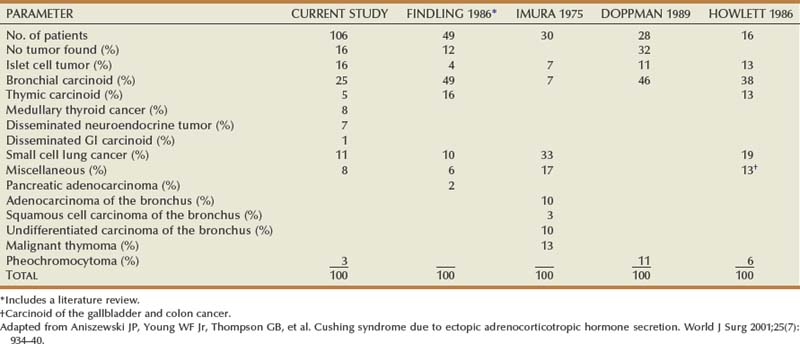
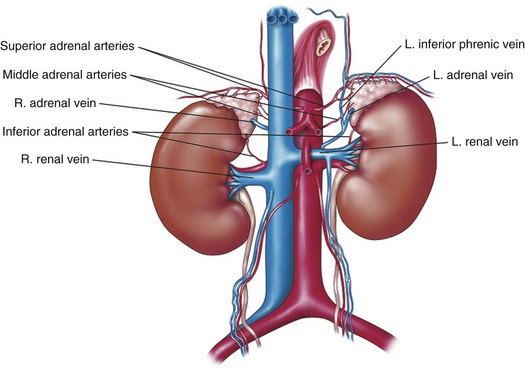
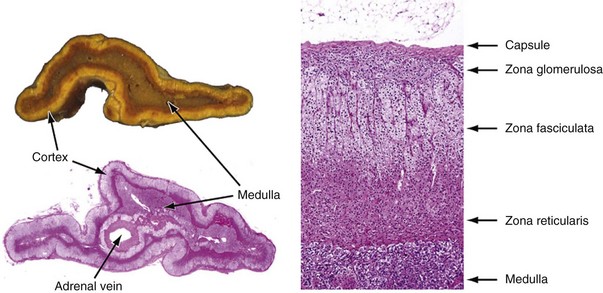
Alexander Kutikov, MD, Paul L. Crispen, MD, Robert G. Uzzo, MD The diminutive adrenal glands were easily overlooked by early physicians. Distinguished anatomists such as Galen, da Vinci, and Vesalius omitted the adrenal glands in their descriptions of the retroperitoneum. Bartholomaeus Eustachius was the first to describe the organs in the mid-16th century (Scott, 1990). Not until the mid-19th century was the critical importance of the adrenal recognized when Thomas Addison, an English physician, described a series of patients with the condition of adrenal insufficiency that now carries his name. He linked the disease to the adrenal glands upon careful inspection of the organs at autopsy (Addison, 1855). Soon thereafter, Charles Brown-Sequard, through a series of animal experiments demonstrated that bilateral adrenalectomy uniformly resulted in death, suggesting that the adrenals were indispensable to the survival of the host (Brown-Sequard, 1856). William Osler was the first to report treatment of Addison disease with hormonal replacement in 1896. He administered crude extract from the adrenal glands of pigs to a patient with Addison disease and produced significant weight gain in this one individual (Oliver and Sharpey-Schafer, 1895). In the ensuing half-century “adrenalin” was discovered, and its production was localized to the adrenal medulla (Oliver and Sharpey-Schafer, 1895). The ability of adrenaline to produce a sustained rise in blood pressure was subsequently determined (Abell and Crawford, 1897). Moreover, the failure of this substance, later termed “epinephrine” to sustain life following bilateral adrenalectomy underscored the complexity and multifunctionality of the adrenal gland and established Addison disease as an ailment of the adrenal cortex (Scott, 1990; Porterfield et al, 2008). Discovery and isolation of cortisol from the adrenal gland in the 1930s and subsequent work on its use to treat rheumatoid arthritis produced a 1950 Nobel Prize in Physiology and Medicine for Edward Kendall, Philip Hench, and Tadeus Reichstein (Scott, 1990). Aldosterone was ultimately isolated from the bovine adrenal in 1952 (Grundy et al, 1952). The latter part of the 20th century witnessed a rapid transformation in our understanding and treatment of adrenal disorders lead by pioneers such as Jerome Conn, Lawson Wilkins, Grant Liddle, and Earl Sutherland (Scott, 1990). The adrenal glands are paired retroperitoneal organs composed of a cortex and medulla. Gross examination reveals a distinct spiculated mustard-yellow–colored cortex that is distinct from the surrounding perinephric fat and the inner medulla, which can be distinguished macroscopically on cross section by its central brown color (see Figs. 57-1 and 57-2 on the Expert Consult website (Courtesy of the University of Kentucky.) (Courtesy of Dr. Thomas J. Sebo, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN.) The adrenal cortex and the medulla are two embryologically and functionally distinct units. The cortex is derived from the intermediate mesoderm of the urogenital ridge (Mitty, 1988; Barwick et al, 2005). Beginning in the 5th week of gestation, mesenchymal cells located at the urogenital ridge and the root of the mesentery proliferate and form the cortex of the fetal adrenal gland. During the 6th and 7th weeks of gestation, additional mesothelial cells surround the fetal adrenal cortex, which will later form the adult adrenal cortex (Mitty, 1988; Barwick et al, 2005). By the end of the 8th week of gestation the mesothelial cells forming the cortex are encapsulated by connective tissue and have separated from the peritoneal mesothelium. In distinction, the adrenal medulla is derived from neural crest cells, located in adjacent sympathetic ganglia, which migrate into the medial aspect of the fetal adrenal cortex by the 9th week of gestation (Kempna and Fluck, 2008). The neural crest cells continue to invade the adrenal cortex until they achieve a central position surrounding the adrenal vein by the 18th week of gestation. This embryologic relationship explains the brown stippling of the adrenal cortex. At birth, the fetal adrenal gland is twice the weight of an adult adrenal gland, but has not completed development. After birth, the fetal cortex begins to atrophy and will be completely resorbed by 12 months of age (Mitty, 1988). As the fetal cortex is being resorbed, the zona glomerulosa and fasciculata of the adult cortex continue to develop, but the zona reticularis will not complete differentiation until 3 years of age, reflecting the relative late importance of sex steroid production by this part of the adrenal cortex (Barwick et al, 2005; Kempna and Fluck, 2008). Cases of unilateral adrenal agenesis are rare and are often associated with unilateral renal agenesis (Nakada et al, 1988; Else and Hammer, 2005). However, because adrenal and renal development are separate processes, this association is likely spurious and is the result of limited radiographic evaluation of the ipsilateral adrenal gland in cases of renal agenesis. Most often, adrenal gland development occurs normally in the absence of ipsilateral renal unit development, malrotation, or malascent. In these cases, the adrenal glands are often discoid in shape and located in their normal position within the retroperitoneum (Mitty, 1988). More common anomalies of adrenal gland development include accessory adrenal tissue and adrenal heterotopia. Accessory renal tissue, also known as adrenal rests, can be composed of cortical or medullary tissues. Due to the close proximity of the adrenal gland and genitourinary organ development, adrenal rests can be found anywhere along the path of gonadal descent within the retroperitoneum. Although adrenal rests can be found in up to 50% of neonates, the tissue typically atrophies and is found in only approximately 1% of adults (Barwick et al, 2005). In cases of congenital adrenal hyperplasia (CAH), adrenal rest within the testis may become hyperplastic and present as a testicular mass. This is an important consideration prior to performing an orchiectomy for a testicular mass in patients with CAH. Adrenal heterotopia results from incomplete separation of primitive adrenal mesoderm from adjacent organs such as the liver or kidney, resulting in partial or complete incorporation of the gland into the adjacent organ. As with most endocrine organs, the blood supply to the adrenal gland is redundant. Although variable, the arterial supply of each adrenal gland may arise from three main sources: superior adrenal arteries (branches from the inferior phrenic arteries), middle adrenal arteries (direct visceral branches from the aorta), and inferior adrenal arteries (branches from the ipsilateral renal artery (see Fig. 57–1). The main adrenal arteries then branch to form a subcapsular plexus. From the subcapsular plexus some branches continue directly to the medulla, while others form sinusoids supplying the adrenal cortex. On the venous side, medullary veins coalesce to form the adrenal vein, which is surrounded by medullary tissue within the adrenal gland. The short right adrenal vein drains directly into the vena cava. On the left, the adrenal vein is long compared with the right and is joined by the inferior phrenic vein prior to draining into the left renal vein. The overlapping of both arterial and venous anatomy makes partial adrenalectomy possible with little risk of subsequent adrenal infarction. The principal lymphatic drainage of the adrenal glands is through the paracaval lymph nodes on the right and the para-aortic lymph nodes on the left. Each adrenal gland is enclosed within a fibrous capsule (see Fig. 57–2). Directly beneath the capsule is the cortex, which comprises three zones: the zona glomerulosa, the zona fasciculata, and the zona reticularis. The zona glomerulosa consists of small polyhedral cells with scant eosinophilic cytoplasm and dark round nuclei. The essential function of the glomerulosa is the production of mineralocorticoids, predominantly aldosterone. The broad layer of large pale cells arranged in vertical columns beneath the zona glomerulosa is the zona fasciculata responsible for the production of glucocorticoids such as cortisol. The zona reticularis, the innermost layer of the cortex, consists of round dark staining cells and predominantly produces sex steroids, such as adrenal estrogens and androgens (Mitty, 1988; MacLennan et al, 2003) (Table 57–1). Table 57–1 Primary Effects of Mineralocorticoids NH4, ammonia. As part of a multistep synthetic pathway, numerous enzymes of the adrenal cortex catalyze conversion of essential steroid hormones from the common precursor cholesterol. Low-density lipoprotein (LDL) serves as the primary source of cholesterol for the adrenal (Arlt and Stewart, 2005). Ratios and types of enzymes in each zone of the adrenal cortex vary, resulting in different hormonal products for each region (Fig. 57–3) (Rainey, 1999). (From Hyun G, Kolon TF. A practical approach to intersex in the newborn period. Urol Clin N Am 2004;31:435–43.) Steroid hormone receptors are absent on cellular membranes of target tissues. Instead, steroids diffuse passively into the cell and bind to their respective receptors intracellularly. Gene transcription is then modulated by direct binding of the hormone receptor complex to target DNA (Arlt and Stewart, 2005). As discussed previously, the three main zones of the adrenal cortex are zona glomerulosa, zona fasciculata, and zona reticularis with each zone primarily responsible for mineralocorticoid (100 to 150 mcg/day), glucocorticoid (10 to 20 mg/day), and androgen synthesis (>20 mg/day), respectively (Arlt and Stewart, 2005). The zona glomerulosa is the outermost region of the adrenal cortex and is the only zone of the adrenal gland that contains the enzyme aldosterone synthase (CYP11B2). As a result, the glomerulosa cells of this tissue are the sole source of aldosterone—the primary human mineralocorticoid (Rainey, 1999). Aldosterone regulates electrolyte metabolism by stimulating epithelial cells of the distal nephron to reabsorb Na+ and Cl−, while secreting H+ and K+. Although aldosterone levels have a profound effects on total body Na+, concentration of the ion does not change, whereas reabsorbtion of sodium is accompanied by reuptake of free water. Therefore aldosterone primarily affects total body volume and not sodium concentration (White, 1994; Arlt and Stewart, 2005). Electrolyte balance in epithelial cells of the submaxillary salivary glands and the large intestine are also under mineralocorticoid control; however, the physiologic importance of this phenomenon is likely minimal (Bastl and Hayslett, 1992). Aldosterone levels are primarily regulated by angiotensin II through the renin-angiotensin-aldosterone system and directly by serum potassium levels. A rise in adrenocorticotropic hormone (ACTH) can also increase aldosterone secretion, but this is a much less potent stimulus (White, 1994; Arlt and Stewart, 2005). For this reason the zona glomerulosa is the only region of the adrenal cortex that does not atrophy upon pituitary failure (Hubbard et al, 1990). Atrial natriuretic peptide is the main inhibitory regulator of aldosterone secretion providing an important link between cardiac, adrenal and renal function; although, somatostatin, dopamine, and others may also play a role (Spat and Hunyady, 2004). A working knowledge of the renin-angiotensin-aldosterone system is critical in conceptualizing and managing hyperaldosteronism. The pathophysiology of this pathway is discussed in the section on Conn syndrome. The zona fasciculata is the site of glucocorticoid production due to zonal expression of 17α-hydroxylase, 21-hydroxylase, and 11β-hydroxylase enzymes (see Fig. 57–3) (Arlt and Stewart, 2005). Cortisol is the primary glucocorticoid in humans, and its secretion is under tight control of the ACTH. As such, cortisol and ACTH are a part of a classic hormonal negative feedback system that features the hypothalamus, the pituitary gland, and the adrenal. The physiology of this axis is discussed in more detail in the section on Cushing syndrome (Jacobson, 2005). Production of cortisol by the adrenal follows a strict circadian schedule with the majority of the hormone being secreted in the early morning (Jacobson, 2005). Glucocorticoids are essential to life and modulate complex physiologic pathways that include metabolism, immunity, maintenance of intravascular volume, regulation of blood pressure, and complex modulation of the central nervous system with significant effects on mood, sleep, and potentially memory (Arlt and Stewart, 2005) (Table 57–2). Table 57–2 Primary Effects of Glucocorticoids From Howards SS, Carey RM. The adrenals. In: Gillenwater JY, Grayhack JT, Howards SS, Duckett JW, editors. Adult and pediatric urology. 2nd ed. Chicago: Year Book; 1991. The zona reticularis is the innermost zone of the adrenal cortex. The presence of 17α-hydroxylase and 17,20-lyase results in the production of dehydroepiandrosterone (DHEA), sulfated DHEA (DHEA-S) and androstenedione to this region, although some androgen synthesis also occurs in the zona fasciculata. Adrenal androgen secretion appears to be under control of ACTH, and, like cortisol, exhibits circadian patterns (Arlt and Stewart, 2005). DHEA, DHEA-S, and androstenedione comprise the greatest portion of steroid hormone that is produced by the adrenals (>20 mg/day), but appear to be the least important for adult physiologic homeostasis (Nguyen and Conley, 2008); however, pharmacologic manipulation of adrenal androgen production remains a viable and increasingly targeted strategy for advanced prostate cancer (see Chapter 133). Moreover, aberrations in production of these hormones during development are responsible for significant pathology and result in the clinical entity known as congenital adrenal hyperplasia (CAH) (Finkelstein and Shaefer, 1979; Hubbard et al, 1990). CAH is discussed in detail in Chapter 110. The adrenal medulla comprises less than 10% of total adrenal mass. Neither its function nor its embryology are related to the neighboring cortex. Instead, this portion of the adrenal, which lies at the center of the gland, is an integral part of the autonomic nervous system. Chromaffin cells of the medulla are innervated by preganglionic sympathetic fibers of T-11 through L-2, making them analogous to cells of the sympathetic ganglia. The medulla secretes epinephrine (80%), norepinephrine (19%), and dopamine (1%). These substances, collectively known as catecholamines, are produced from the amino acid tyrosine and modulate the systemic stress response (Fig. 57–4) (Robertson, 1990; de Diego et al, 2008). The effects of these catecholamines are mediated through their binding to adrenoreceptors located on target organs. The nature of these effects depend on the adrenoreceptor types or subtypes located and stimulated on a particular end organ (Table 57–3) (Robertson, 1990; Guimaraes and Moura, 2001). Interestingly, the enzyme phenylethanolamine-N-methyl transferase (PNMT), which catalyzes the conversion of norepinephrine to epinephrine, is relatively unique to the adrenal medulla (the brain and organ of Zuckerkandl also express this enzyme). The function of this enzyme is potentiated by the presence of glucocorticoids, thereby creating one of the few physiologic links between the adrenal cortex and the medulla. Localization of PNMT to the adrenal medulla explains why the gland is the primary source of systemic epinephrine, despite the presence of similar chromaffin cells elsewhere in the sympathetic nervous system (Robertson, 1990). Similar to the physiology that controls norepinephrine release at synaptic nerve terminals, the storage and release of adrenal catecholamines involves intracellular vesicles. Liberation of these vesicles through exocytosis results in release of adrenal catecholamines into the blood stream (Eisenhofer et al, 2004b). The metabolism of catecholamines is complex and some controversy still exists about the physiologic relevance of each pathway (Eisenhofer et al, 2004b). The majority of adrenal catecholamine metabolism occurs at the scene of production in cells of the adrenal medulla themselves (Eisenhofer et al, 2003a). For clinical purposes, three metabolites (metanephrine, normetanephrine, and vanillylmandelic acid) and two enzymes (catechol-O-methyltransferase and monoamine oxidase) are important. Metanephrine and normetanephrine result from respective methylation of epinephrine and norepinephrine by catechol-O-methyltransferase (COMT). Although large amounts of this enzyme are present in the liver and kidneys, the majority of adrenal catecholamine metabolites are methylated by COMT within the cells of the adrenal medulla (Eisenhofer et al, 2003a). Indeed, over 90% of metanephrine (epinephrine metabolite) and some 20% or more of normetanephrine (a norepinephrine metabolite) in the blood stream are derived from the adrenal medulla. Therefore a measurable rise in the level of these metabolites is very useful when diagnosing potential pheochromocytomas (see section on Assessment of Function of Adrenal Masses) (Eisenhofer et al, 1995). In the urine, the majority of these metabolites are excreted in a sulfonated form. Furthermore, monoamine oxidase (MAO) and other enzymes subsequently participate in the conversion of catecholamine metabolites to vanillylmandelic acid (VMA), the primary catecholamine metabolic end product that is largely formed by the liver. Nonadrenal catecholamines from the sympathetic nervous system are similarly converted to VMA (Eisenhofer et al, 2004b). Figure 57–4 summarizes the most clinically relevant aspects of the catecholamine metabolism pathway. Hypercortisolism secondary to excessive production of glucocorticoids by the adrenal cortex is defined as Cushing syndrome (Orth and Liddle, 1971). The disease is rare and occurs in 2 to 5 of every million people per year (Lindholm et al, 2001; Findling and Raff, 2005). Diagnosis and treatment of Cushing syndrome is multifaceted, often requiring cooperation of internists, endocrinologists, neurosurgeons, and adrenal surgical experts (Newell-Price et al, 2006). The urologist must understand the comprehensive pathophysiology of hypercortisolism and have particularly advanced expertise in the aspects of this complex disease for which adrenalectomy is required. The zona fasciculata of the adrenal cortex secretes more than 20 mg of cortisol every day (Arlt and Stewart, 2005). Regulation of this secretion is controlled through the hypothalamic-pituitary-adrenal (HPA) axis (Jacobson, 2005). A fluent understanding of this classic neuroendocrine negative feedback system is critical for the successful management of every Cushing patient. Corticotrophic cells of the anterior pituitary secrete ACTH, also known as corticotrophin, under the influence of neuronal innervations from the hypothalamus. Synthesis of ACTH is a result of cleavage of the pro-opiomelanocortin (POMC) precursor molecule. Physiologically, the most important promoter of ACTH release is corticotropin-releasing hormone (CRH), but oxytocin and vasopressin also play a role (Jacobson, 2005; Sam and Frohman, 2008). Stress, whether physiologic or psychologic, appears to be the most important variable in modulating activity of the HPA axis (Jacobson, 2005). ACTH is not only responsible for promoting production of glucocorticoids and androgens by the adrenal cortex, but it also plays a critical role in maintaining adrenal cortical vitality. Indeed, without ACTH (e.g., when its secretion is suppressed by exogenous steroid intake), all but the mineralocorticoid-producing cells of the adrenal cortex arrest hormone production and undergo apoptosis (Arlt and Stewart, 2005; Jacobson, 2005). In addition to ACTH, splanchnic nerves that innervate the adrenals also appear to affect glucocorticoid production (Jacobson, 2005). Glucocorticoids bind receptors in the hypothalamus and the pituitary and complete the negative feedback loop by inhibiting production of CRH and ACTH by these structures, respectively (Jacobson, 2005). Figure 57–5 summarizes the salient features of the HPA axis. It is important to understand that CRH secretion is under tight control of the hypothalamic suprachiasmic nucleus and follows circadian patterns. The highest level of cortisol in healthy subjects is detected in the mornings, while the nadir is observed at approximately 11 PM. Even small perturbations of this physiologic rhythm are considered pathologic (Jacobson, 2005; Sam and Frohman, 2008). Given the sophistication of the HPA axis, hypercortisolism can result from a number of different pathologies that result in oversecretion of cortisol by the adrenal glands. Causes of Cushing syndrome can be divided into three main groups: (1) exogenous, (2) ACTH-dependent, and (3) ACTH-independent (Fig. 57–6). Exogenous Cushing syndrome is a result of iatrogenic glucocorticoid administration. ACTH-dependent disease results from an elevated serum corticotropin level due to pathology extrinsic to the adrenal gland and accounts for up to 85% of cases of endogenous Cushing syndrome. ACTH-independent hypercortisolism, on the other hand, results from unregulated overproduction of glucocorticoids by the adrenal(s) and is relatively rare. The adrenal surgeon has long been involved in management of ACTH-independent disease, but with advent of laparoscopy, adrenal surgery for ACTH-dependent Cushing syndrome is becoming more commonplace (Porterfield et al, 2008). Exogenous Cushing syndrome is the most common cause of hypercortisolism in patients of the Western world (Newell-Price et al, 2006). Synthetic glucocorticoids are frequently employed in treatment of many conditions, and Cushing syndrome can result even from the administration of even low doses of exogenous steroids administered orally, topically, or by inhaled preparations (Hopkins and Leinung, 2005; Newell-Price et al, 2006). A careful patient history is therefore essential in the evaluation of all patients with Cushing syndrome. The clinician also must be cognizant of the possibility that the patient is either unaware of his or her steroid use (e.g., use of herbal remedies, nasal sprays) or is surreptitiously self-administering glucocorticoids (e.g., for performance enhancement) (Hopkins and Leinung, 2005). ACTH-dependent hypercortisolism accounts for 80% to 85% of endogenous Cushing syndrome. Approximately 80% of ACTH-dependent disease is due to primary pituitary pathology and is known as Cushing disease. Ectopic ACTH production is the other main cause of ACTH-dependent hypercortisolism (Newell-Price et al, 2006). Ectopic CRH syndrome is a third cause of ACTH-dependent Cushing syndrome and is extremely uncommon (see Fig. 57–6) (Orth, 1995). Described by the neurosurgical pioneer Harvey Cushing in 1932, Cushing disease causes hypercortisolism through excessive secretion of ACTH by the pituitary gland (Cushing, 1932). The condition accounts for nearly 70% of endogenous Cushing syndrome. The most common culprit is a corticotropin-producing microadenoma. Large macroadenomas of 1 cm in size or greater are found in only about 5% of cases (Newell-Price et al, 2006). Fifteen percent of functional pituitary adenomas exhibit the ability to secrete ACTH (Shimon and Melmed, 1998). Hyperplasia of ACTH-producing cells and pituitary carcinoma can also oversecrete ACTH but are extremely rare (Orth, 1995). Up to two thirds of individuals with Cushing disease are female (Scott and Orth, 1990). Production of ACTH by nonpituitary tumors can also result in hypercortisolism. These corticotropin-producing tissues are nearly always malignant and account for approximately 10% of Cushing syndrome (Porterfield et al, 2008). Liddle and associates first described the phenomenon in the early 1960s and termed it “ectopic ACTH syndrome” (Meador et al, 1962; Scott and Orth, 1990). The timing and onset of hypercortisolism varies and can precede the diagnosis of an extra-adrenal malignancy by many years, resulting in diagnostic difficulties and the clinical conundrum, whereby a subclinical pituitary adenoma is suspected but not radiographically identified (Aniszewski et al, 2001). Moreover, hypercortisolism in end-stage debilitated cancer patients is almost certainly underdiagnosed (Orth, 1995). As a result of higher ACTH levels, the adrenal glands of patients with ectopic ACTH syndrome are generally more markedly hyperplastic and thereby larger than those of patients with Cushing disease (Jenkins et al, 1999). Table 57–4 summarizes primary malignancies implicated in ectopic ACTH production, which notably includes pheochromocytoma. Production of CRH by malignancies is extremely rare and is responsible for less than 1% of cases of Cushing syndrome. Bronchial carcinoma is the most common culprit, and concurrent ACTH secretion is not unusual (Carey et al, 1984; Orth, 1995). Uncontrolled hypersecretion of cortisol by adrenal tissues accounts for a minority of endogenous hypercortisolism and is classified as ACTH-independent Cushing syndrome. Adrenal neoplasms and rare forms of bilateral adrenal disease are responsible for this group of conditions (Lacroix and Bourdeau, 2005; Newell-Price et al, 2006). Cortisol-secreting benign adenomas of the adrenal gland are responsible for approximately 10% of Cushing syndrome and often result from a dominant unilateral hyperplastic nodule, although multifocal bilateral functional adrenal hyperplasia may also occur. Less than 10% of adrenal masses are bilateral and often present a diagnostic challenge (Lacroix and Bourdeau, 2005; Porterfield et al, 2008; Young et al, 2008). Radiographically undetectable benign adrenal cortisol-producing lesions may also be responsible for cases of subclinical Cushing syndrome (Rossi et al, 2000). Cortisol production by adrenocortical carcinomas account for approximately 8% of patients with Cushing syndrome (Lindsay and Nieman, 2005a). ACTH-independent bilateral macronodular adrenal hyperplasia (AIMAH) gives rise to less than 1% of Cushing syndrome. The condition is characterized by multiple large (up to 4 cm) nodules replacing the glands with each adrenal weighing over 60 g (Lacroix and Bourdeau, 2005; Iacobone et al, 2008). Weights in excess of 200 g have been reported for each gland affected by AIMAH (Swain et al, 1998). It is essential that this disease entity must be distinguished from adrenal enlargement caused by chronic adrenal stimulation in ACTH-dependent Cushing syndrome, whereas radiographics of the adrenals appear similar in both conditions. Bilateral macronodular hyperplasia is observed as a feature of the McCune-Albright syndrome, which also includes polyostotic fibrous dysplasia, dermatologic manifestations, and other endocrine abnormalities (Lacroix and Bourdeau, 2005). Like AIMAH, primary pigmented nodular adrenocortical disease (PPNAD) is exceedingly rare, accounting for less than 1% of cases of Cushing syndrome. Unlike AIMAH, however, the adrenal glands in this condition remain normal in size and exhibit black or brown cortical nodules (Young et al, 1989). The cortical tissue surrounding the nodules is atrophic and the adrenal medulla is free of disease (Lacroix and Bourdeau, 2005). Approximately half of PPNAD is found in patients with the autosomal-dominant Carney complex, which is also responsible for spotty skin and mucous membrane lesions, and a variety of neoplasms that include Sertoli-cell tumors. The other half of cases of PPNAD are nonhereditary with no known cause (Lacroix and Bourdeau, 2005). Clinical characteristics of Cushing syndrome vary considerably among affected patients. Many of the classic symptoms of hypercortisolism, such as central obesity, moon faces, buffalo hump, proximal muscle weakness, easy bruisability, and abdominal striae, are non-specific. It is the combination of these and other clinical signs that should raise suspicion and prompt potential screening for Cushing syndrome (Findling and Raff, 2005). Cushing syndrome also results in systemic symptomatology, such as central obesity, dyslipidemia, insulin resistance, and hypertension, that is identical to the highly-prevalent metabolic syndrome (Pivonello et al, 2005, 2008). Figure 57–7 depicts the effects of glucocorticoids on organ systems, and Table 57–5 summarizes signs and symptoms of the disease and indicates their frequency. Screening for Cushing syndrome is usually beyond the scope of urologic practice; however, it is important for urologists to appreciate the relatively common occurrence of hypogonadal hypogonadism in men with Cushing syndrome. A low threshold for initiating a hypercortisolism workup should exist in these men with libido or erectile problems, low testosterone, and low gonadotropin levels (Findling and Raff, 2005; Pivonello et al, 2008). Up to 50% of patients with Cushing syndrome exhibit urolithiasis; therefore stone formers with cushinoid features also deserve a hypercortisolemia evaluation. Interestingly, definitive treatment of Cushing syndrome in these patients reduces the risk of stone formation, but does not bring the risk back to that of the general population (Faggiano et al, 2003). Table 57–5 Signs and Symptoms of Cushing Syndrome Adapted from Pivonello R, De Martino MC, De Leo M, et al. Cushing’s syndrome. Endocrinol Metab Clin North Am 2008;37(1):135–49, ix. Subclinical Cushing syndrome is a relatively new disease entity that describes hypercortisolemia in the absence of an overt cushinoid phenotype. Obvious clinical signs of Cushing syndrome are absent, and the diagnosis is made when a metabolic workup for an incidentally-discovered adrenal mass reveals hypercortisolemia (Sippel and Chen, 2004). In the past, the disease entity was referred to as “pre-Cushing syndrome,” “noncushingoid Cushing syndrome,” and “preclinical Cushing syndrome”; however, the term “subclinical Cushing” has gained acceptance. Recently, the term “subclinical autonomous glucocorticoid hypersecretion” also has been introduced (Tsagarakis et al, 2006). Classically, the disease entity applies to patients who are found to have elevated cortisol levels upon a metabolic workup of an adrenal incidentaloma. Nearly 10% of patients in one large series of adrenal incidentalomas were found to have the condition (Mantero et al, 2000). Patients can also fall under the umbrella of this diagnosis when Cushing syndrome is discovered during screening. For instance, when patients with type II diabetes and poor glucose control are screened for hypercortisolism, some 2% have subclinical Cushing syndrome (Catargi et al, 2003). Adrenalectomy in the setting of subclinical Cushing may improve glucose control, hypertension, and result in weight loss (Midorikawa et al, 2001; Mitchell et al, 2007). Surgical indications and benefits for subclinical Cushing syndrome, however, are still a matter of debate (Sippel and Chen, 2004; Tsagarakis et al, 2006). Some authors argue that adrenalectomy should be performed only in patients who are potentially symptomatic and exhibit clinical signs, such as hypertension, obesity, glucose intolerance, or osteopenia. Others propose that surgery must be offered to all patients in order to prevent the sequelae of hypercortisolism (Sippel and Chen, 2004; Pivonello et al, 2005; Tsagarakis et al, 2006). Patients with subclinical Cushing syndrome may be at higher risk for postadrenalectomy adrenal insufficiency than those patients with non-cortisol-secreting adrenal pathologies, because functionality of the contralateral gland may be suppressed (Tsagarakis et al, 2006). The diagnosis of Cushing syndrome is one of the most complex and difficult tasks in clinical endocrinology and most often falls outside of the realm of medical or surgical urological practice (Findling and Raff, 2005). Nevertheless, the urologist must be familiar with these diagnostic strategies and how they relate to the etiology of hypercortisolism. Moreover, the urologist who offers adrenalectomy must be well versed in the basic metabolic evaluation for Cushing syndrome in patients with adrenal masses. When the diagnosis of Cushing syndrome is suspected, the clinician can choose among several laboratory studies. The two evaluations performed most frequently are the 24-hour urinary free cortisol evaluation and the overnight low-dose dexamethasone suppression test (LD-DST). The sensitivity of the former test, however, may not be adequate for screening patients with incidentalomas (Nieman et al, 2008). Late-night salivary cortisol is a test that is becoming increasingly popular. Second-line tests include the 2-day LD-DST and midnight plasma cortisol testing (Arnaldi et al, 2003; Newell-Price et al, 2006, Nieman et al, 2008; Pivonello et al, 2008). The physiologic principles underlying these tests are important to understand and are described in this section (Findling and Raff, 2005). The section Evaluation of Adrenal Pathology in Urologic Practice outlines practical algorithms for the metabolic evaluation of adrenal lesions. Tests such as random serum cortisol, plasma ACTH level, urinary 17-ketosteroid level, insulin tolerance testing, and loperamide testing have fallen out of favor and are no longer recommended (Nieman et al, 2008). Administration of low-dose dexamethasone followed by measurement of serum cortisol the next morning probes the patient’s glucocorticoid negative feedback system. In subjects without hypercortisolism, dexamethasone acts on the corticotrophic cells of the anterior pituitary, suppresses ACTH production, and thereby results in a reduction of serum cortisol levels (Findling and Raff, 2005). A key element to understanding this test is that despite the “low-dose” designation, the dosage of dexamethasone administered for this study corresponds to three- to fourfold levels of physiologic glucocorticoid. Importantly, the exogenous dexamethasone is not detected by the serum cortisol assay. The fact that patients with ACTH-independent Cushing syndrome and those with ectopic ACTH secretion fail to suppress cortisol production during such testing is intuitive; however, the reason for the failure of patients with ACTH-producing pituitary adenomas (Cushing disease) to suppress cortisol secretion following low-dose dexamethasone administration is less obvious. The phenomenon is best understood by the relative insensitivity of pituitary adenomas to the inhibitory effects of glucocorticoid stimulation (Raff and Findling, 2003). In summary, a patient’s failure to suppress cortisol levels following an overnight low-dose dexamethasone administration is indicative of Cushing syndrome. This test does not delineate the etiology of hypercortisolism, but simply suggests its presence. Administration of low-dose dexamethasone over a 48-hour period is less practical and is reserved for second-line testing. Urinary free cortisol evaluation is a 24-hour direct measurement of free bioavailable cortisol. Unlike serum cortisol testing, which measures both free and bound cortisol, urinary cortisol measurements are independent from the variables that influence corticosteroid-binding globulin levels (Arnaldi et al, 2003). Moreover, the test is an integrated measurement of cortisol secretion over a 24-hour period. This feature of the study is critical, because serum cortisol levels exhibit some circadian variation even in many patients with Cushing syndrome (Orth, 1995). The urologist must be cognizant that this test may not possess sufficient sensitivity for diagnosis of subclinical Cushing syndrome, and the Endocrine Society recommends against it for metabolic evaluation of adrenal incidentalomas (Nieman et al, 2008). Late-night salivary cortisol and midnight plasma cortisol measurements take advantage of a common feature of all causes of Cushing syndrome—a perturbation and in some cases complete disruption in the diurnal variation of cortisol levels. The abnormality, even in very mild cases of Cushing syndrome, is the inability to suppress cortisol levels at night. As previously mentioned, peak morning cortisol levels in patients with Cushing syndrome are often within the normal range; however, persistent elevation at night may signal the loss of diurnal variance associated with Cushing. While midnight plasma cortisol measurements are clinically impractical in an outpatient setting, late-night salivary cortisol measurements are becoming a popular diagnostic tool for identification of hypercortisolism (Raff and Findling, 2003; Findling and Raff, 2005). It is important to note that other conditions can stimulate the hypothalamic-pituitary-adrenal axis and emulate Cushing syndrome (Table 57–6); therefore endocrinologic expertise is recommended when definitively diagnosing hypercortisolism (Nieman et al, 2008). Table 57–6 Causes of Hypercortisolism in the Absence of Cushing Syndrome Whereas Cushing syndrome is unlikely in these conditions, it may rarely be present. If there is a high clinical index of suspicion, the patient should undergo testing, particularly those within the first group. Adapted from Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008;93(5):1526–40. Once Cushing syndrome is diagnosed, its etiology must be localized. Due to the nuanced complexity of this process, some authors believe that this is best done by endocrinologists at major referral centers (Newell-Price et al, 2006). Briefly, the algorithm is as follows. First, ACTH-dependent disease must be distinguished from the ACTH-independent causes. This is done by measurement of serum ACTH. Low serum ACTH points to ACTH-independent pathology, and abdominal imaging is indicated to identify the adrenal source. If the adrenals are unremarkable on cross-sectional imaging, exogenous steroids as a cause of Cushing syndrome, or much less commonly PPNAD, must be suspected (Arnaldi et al, 2003
Historical Background
Adrenal Anatomy and Embryology
Overview
 ). The normal adrenal gland weighs an average of 4 to 5 g each and range in size from 4 to 6 cm in length and 2 to 3 cm in width (Mitty, 1988; Silverman and Lee, 1989; Avisse et al, 2000). Morphologically, the right adrenal gland is triangular in shape while the left adrenal gland is crescent shaped (Avisse et al, 2000). They may sit either immediately superior to the kidney, “capping” the upper pole, or superior medially to the upper pole, “cradled” by the kidney just above the renal vessels. Once understood, this difference can be appreciated by either computed tomography (CT) scan or magnetic resonance imaging (MRI) and is a relevant surgical distinction, particularly in patients with substantive perinephric fat and on the left side when identifying the adrenal vein.
). The normal adrenal gland weighs an average of 4 to 5 g each and range in size from 4 to 6 cm in length and 2 to 3 cm in width (Mitty, 1988; Silverman and Lee, 1989; Avisse et al, 2000). Morphologically, the right adrenal gland is triangular in shape while the left adrenal gland is crescent shaped (Avisse et al, 2000). They may sit either immediately superior to the kidney, “capping” the upper pole, or superior medially to the upper pole, “cradled” by the kidney just above the renal vessels. Once understood, this difference can be appreciated by either computed tomography (CT) scan or magnetic resonance imaging (MRI) and is a relevant surgical distinction, particularly in patients with substantive perinephric fat and on the left side when identifying the adrenal vein.
Embryology
Anatomy
Histology
ACTION
EFFECT
SITE OF ACTION
Renal sodium reabsorption
Renal chloride reabsorption
Renal potassium secretion
Renal proton secretion
Adrenal Physiology
Adrenal Cortex Physiology
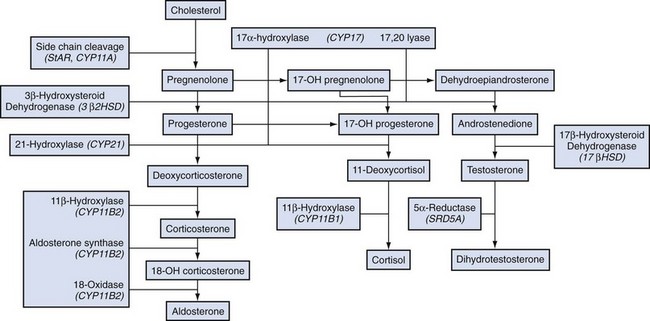
Zona Glomerulosa
Zona Fasciculata
EFFECTS
CLINICAL IMPLICATIONS
Enhance skeletal and cardiac muscle contraction
Absence results in weakness
Cause protein catabolism
Excess results in muscle wasting and weakness
Inhibit bone formation
Excess decreases bone density
Inhibit collagen synthesis
Excess causes thin skin and fragile capillaries
Increase vascular contractility and decrease permeability
Absence makes it difficult to maintain blood pressure
Have anti-inflammatory activity
Exogenous steroid is useful in treating inflammatory diseases
Have anti-immune system activity
Exogenous steroids are useful in promoting transplant tolerance and in treating autoimmune disorders
Maintain normal glomerular filtration (GFR)
Absence reduces glomerular filtration rate
Zona Reticularis
Adrenal Medulla Physiology
Adrenal Disorders
Disorders of Increased Adrenal Function
Cushing Syndrome
Introduction and Epidemiology
Pathophysiology
Introduction
Normal Hypothalamic-Pituitary-Adrenal (HPA) Axis Physiology
Overview of Cushing Syndrome
Exogenous Cushing Syndrome
ACTH-Dependent Cushing Syndrome
Overview
Cushing Disease
Ectopic Acth Syndrome
Ectopic CRH
Acth-Independent Cushing Syndrome
Adrenal Tumors
Acth-Independent Macronodular Adrenal Hyperplasia (AIMAH)
Primary Pigmented Nodular Adrenocortical Disease (PPNAD)
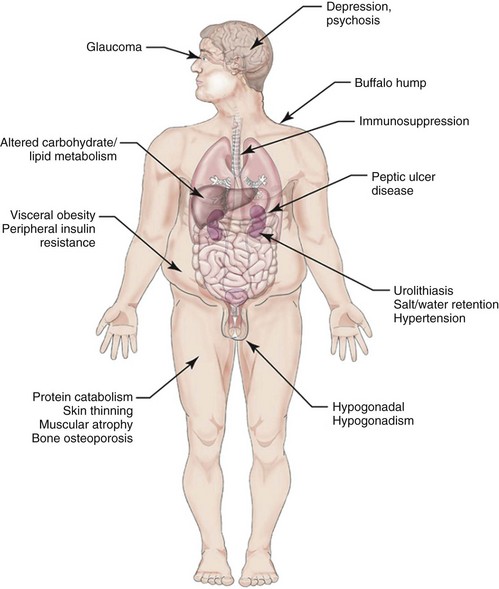
Clinical Characteristics
Classical Cushing Syndrome
SYMPTOMS AND SIGNS
PREVALENCE (%)
Central obesity
90-100
Rounded face (“moon face”)
Facial plethora
Decreased libido
Purple striae
70-90
Menstrual disturbances
Hirsutism
Erectile dysfunction
Hypertension
Muscle weakness
50-70
Posterior neck fat deposit (“buffalo hump”)
Body bruising
Glucose intolerance/diabetes
Osteopenia/osteoporosis
Emotional lability/depression
Headache
20-50
Backache
Limb edema
Recurrent infections
Hypokalemic alkalosis
Nephrolithiasis
Acne
0-20
Alopecia
Subclinical Cushing Syndrome
Diagnostic Tests
Establishing the Diagnosis of Cushing Syndrome
Conditions
Identifying the Cause of Cushing Syndrome
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree