Malignant renal cell tumours
Clear cell renal cell carcinoma
Multilocular clear cell renal cell carcinoma
Papillary renal cell carcinoma
Chromophobe renal cell carcinoma
Carcinoma of the collecting ducts of Bellini
Renal medullary carcinoma
Xp11 translocation carcinomas
Carcinoma associated with neuroblastoma
Mucinous tubular and spindle cell carcinoma
Renal cell carcinoma unclassified
The pathology of familial renal cancers is summarised in Table 19.2.
Table 19.2
Pathology of hereditary RCC
Syndrome | Chromosomes involved | Gene | Protein | Histological type | Involvement of other organs |
|---|---|---|---|---|---|
Von Hippel-Lindau | 3p25 | VHL | pVHL | Bilateral, cystic renal lesions and clear cell | Cysts in other organs, phaeochromocytoma neuroendocrine tumours |
Carcinoma | |||||
Hereditary papillary Ca | 7p 31 | c-MET | HGF-R | Bilateral & multiple | None |
Papillary RCC | |||||
HLRC (Hereditary leiomyomatosis RCC | 1q 42 | FH | FH | Papillary RCC (non type 1) | Uterine leiomomyoma/leiomyosarcoma |
Familial papillary thyroid carcinoma | 1q 21 | Not known | Not known | Papillary RCC & oncocytoma | |
Hyperparathyroidism jaw tumour (HP-JT) | 1q 25 | HRPT2 | – | Epithelial stromal mixed tumours, papillary RCC | |
Birt-Hogg-Dube | 17p 11 | BHD | Folliculin | Multiple chromophobe RCC, oncocytic adenoma, papillary RCC | Pulmonal cysts; spontaneous |
Pneumothorax, facial fibrofolliculoma | |||||
Constitutional translocation chromosome 3 | 3p13-14 | Multiple, bilateral clear cell RCC | – |
Table 19.3 outlines rare tumours in the kidney in WHO classification [5].
Table 19.3
Benign and other rare malignant tumours
1. Benign: papillary adenoma, oncocytoma |
2. Metanephric tumours (metanephric adenoma, metanephric adenofibroma, metanephric stromal tumours) |
3. Nephroblastic tumours (cystic nephroma, mixed epithelial and stromal tumours, synovial sarcoma) |
4. Nephroblastic tumours (nephrogenic rests, nephroblastoma, cystic partially differentiated nephroblastoma) |
5. Neuroendocrine tumours (carcinoid, neuroendocrine carcinoma, primitive neuroectodermal tumour, Neuroblastoma, pheochromocytoma) |
6. Miscellaneous (mesenchymaltumours, haemopoetic & lympoidtumours, germ cell tumours, metastatictumours) |
Table 19.4 summarizes the grouping of the tumours according to the prognosis: good prognosis, intermediate and poor prognosis.
Table 19.4
Clinical classification according to prognosis
Good prognosis | Papillary adenoma (benign); oncocytoma (benign) |
Metanephric adenoma; angiomyolipoma | |
Multilocular clear cell renal cell carcinoma (malignant) | |
Mucinous tubular and spindle cell carcinoma (malignant) | |
Intermediate prognosis | Papillary carcinoma (malignant), chromophobe renal cell carcinoma |
Poor prognosis | Clear cell carcinoma, metastatic cancers, collecting duct carcinoma |
Renal medullary carcinoma, Xp11 translocation carcinomas | |
Sarcomas |
Clear Cell RCC (ccRCC)
The cells of ccRCC have a clear cytoplasm due to the high content of glycogen and lipid but may have an eosinophilic or granular appearance when the mitochondrial content is high (Fig. 19.1). The ccRCC accounts for more than 60 % of all malignant renal tumours with 4 % being multifocal and 3 % bilateral [4]. Compared to other RCCs, they tend to be more aggressive in men, progress to metastatic disease twice as frequently after nephrectomy and have lower overall survival [6].
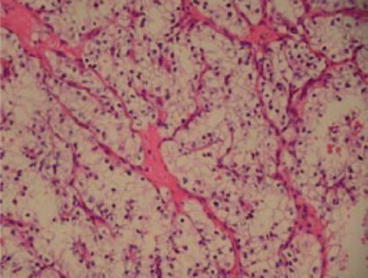
Fig. 19.1
Clear cell carcinoma (×100)
Macroscopically, the cut surface has yellowish appearance often with areas of haemorrhage, scarring and calcification, ossification and extension to renal vein. They maybe solid, tubular or cystic, the latter of which has given rise to a subtype known as multilocular cystic RCC when the there is a complete cystic appearance. Sarcomatoid changes are seen in 5 % of tumours, which represents high-grade transformation and carries a poor prognosis.
Papillary RCC (PRCC)
These are the second most common type, with a male predominance and accounting for up to 15 % of all RCCs [7] (Fig. 19.2).
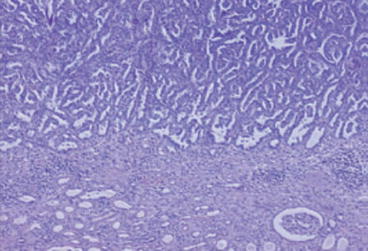
Fig. 19.2
Papillary RCC (×100)
Tumours less than 5 ml are considered to be adenomas and those greater than 5 ml to be carcinomas [7]. The underlying genetic abnormalities are trisomy of chromosomes 3q, 7, 8, 12, 16, 17 and 20 with loss of the Y chromosome [8]. Macroscopically, they are round and white or beige in colour and may contain central necrosis or haemorrhagic areas. The cells are usually small containing scanty cytoplasm and foamy macrophages. The growth pattern is predominantly papillary but may also be tubular or solid. Two morphological types are typically described, although recently a third group has emerged composed entirely of oncocytes and shows different clinic-pathological features [9].
Type I
The papillae and tubular structures are lined by cuboidal cells with small oval nuclei, inconspicuous nucleoli and pale cytoplasm. The papillary cores often contain foamy macrophages and psammoma bodies are common. These patients have a longer survival.
Type II
The papillae contain large cells with higher nuclear grade, copious eosinophilic cytoplasm and large nuclei with prominent nucleoli. Foamy macrophages within the papillary cores and psammoma bodies are uncommon. In general, the type 2 tumours are larger and present in younger patients with advanced stage.
Type III
Oncocytic PRCC – Rarely, PRCC can display extensive areas of solid and non-papillary architecture and extensive areas with oncocytic cytoplasm. The oncocytic features in PRCC may pose a diagnostic problem in differentiating oncocytic PRCC from oncocytoma. Papillae and trabeculae are present with fibro-vascular cores covered with cells containing an eosinophilic cytoplasm packed with mitochondria and aggregates of foamy macrophages, round nuclei and a low nuclear-cytoplasmic ratio. There are no cytogenetic or molecular tests for this variant of PRCC [10].
Chromophobe RCC (CRCC)
They account for 5 % of RCC and are characterised by a combination loss of chromosomes 1, 2, 6, 10, 13, 17 and 21 [4]. Macroscopically, the tumour appears lobulated; the cut surface containing one or more solid tumour nodules but well circumscribed. The cells are large and pale and polygonal usually with a clear but sometimes eosinophilic cytoplasm containing condensed, hyperchromatic and sometimes bifid nuclei. The growth pattern is typically solid although rarely maybe cribiform (Fig. 19.3).
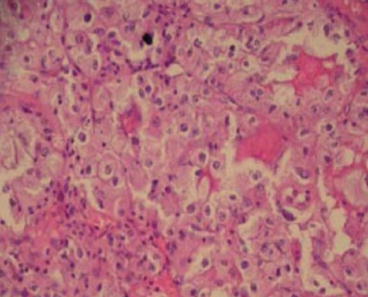
Fig. 19.3
Chromophobe RCC
Rare Types of Renal Cell Carcinomas
Multilocular Clear Cell RCC (MCRCC)
This accounts for an estimated 3.5 % of RCCs and whilst it may be considered as a separate entity it is essentially a well-differentiated ccRCC [7]. The cysts are lines by a single layer of clear cells with fibrous septations containing epithelial cells [7]. They present as a Bosniak type II or III cystic lesion although this type of Bosniak lesion can also be due to a benign cystic nephroma, multilocular cyst or mixed epithelial and stromal tumor of the kidney. Biopsy or frozen section of the lesion often cannot distinguish between these lesions and the usual management is surgical excision. The clinical outcome in these cases is excellent.
Collecting Duct (of Bellini) Carcinoma (CDC)
These rare tumours are derived from the cells of collecting ducts in the renal medulla and account for less than 1.8 % of RCCs [11]. There is a male predominance and poor prognosis with up to 40 % having metastatic disease on clinical presentation [12]. The most frequent genetic abnormality is deletion of chromosome 1q32 [13]. Macroscopically, they have a whitish grey appearance with a solid growth pattern. There is dilation of the tubules and papillae, which are lined by cuboidal cells, giving a cobblestone appearance under microscopy. At presentation they are usually advanced with metastasis with Fuhrman grade 3 or 4 nuclear features (Fig. 19.4 [14], Fig. 19.5).
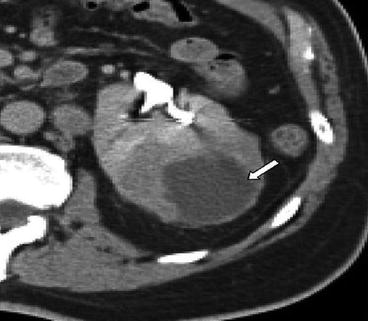 < div class='tao-gold-member'>
< div class='tao-gold-member'>
 Diet and GU Cancers
Diet and GU Cancers
 Life After Urological Cancer – Psychological Issues
Life After Urological Cancer – Psychological Issues
 Genetics and Genito-Urinary Cancer
Castrate Resistant Prostate Cancer: Systemic Chemotherapy and a System Problem
Genetics and Genito-Urinary Cancer
Castrate Resistant Prostate Cancer: Systemic Chemotherapy and a System Problem
 Renal Failure, Dialysis and Transplantation: In the Management of Renal Cell Carcinoma (Solitary Kidney and Bilateral Renal Tumours)
Renal Failure, Dialysis and Transplantation: In the Management of Renal Cell Carcinoma (Solitary Kidney and Bilateral Renal Tumours)
 Normal Cell
Normal Cell




Only gold members can continue reading. Log In or Register to continue
Related posts:
Diet and GU Cancers
Life After Urological Cancer – Psychological Issues
Genetics and Genito-Urinary Cancer
Castrate Resistant Prostate Cancer: Systemic Chemotherapy and a System Problem
Renal Failure, Dialysis and Transplantation: In the Management of Renal Cell Carcinoma (Solitary Kidney and Bilateral Renal Tumours)
Normal Cell
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
