Fig. 3.1
Clonal evolution drives tumor progression. (a) Healthy epithelium. (b) The first oncogenic mutation occurs in a single epithelium cell (Yellow cell) resulting in the growth of a genetically homogeneous benign lesion. (c) A second mutation occurs in one of the cells in the benign lesion (orange cell) conferring a further growth advantage. (d) Increased numbers of rapidly dividing cells increases the likelihood of further mutations (red cell) which leads to (e) the growth of a more malignant and invasive clone within the primary tumor. (f) A further mutation in a cell within the malignant subclone causes further transformation and (g) the capacity for distant metastasis
Within the tumour cell population it is common to find cancer stem cells [2], cells that have many of the traits of normal stem cells and have the capacity to seed new tumours [3]. To date it is not clear how these cancer stem cells originate; however they play a significant role in the pathogenesis of many cancers and may be the primary cause of disease recurrences following radio- and chemo-therapy [4].
Regulators of Tumour Development: Tumour Suppressor Genes and Oncogenes
Genomic alterations that drive tumour growth alter the activity of genes that express proteins responsible for the tight regulation of the cellular pathways involved in DNA replication, growth, differentiation, motility and cell survival (see below in “Molecular genetics”). Genes that express proteins that negatively regulate these processes are termed tumour suppressor genes and genes that positively regulate these processes are termed proto-oncogenes.
Tumour suppressor genes are commonly de-activated during tumour development and inactivation can occur by a variety of mechanisms including mutation, gross chromosomal deletion or epigenetic mechanisms such as promoter methylation.
Proto–oncogenes are normal genes that are necessary for normal cell survival. However mutations may occur that result in these genes being abnormally activated (e.g. mutations may cause activation in the absence of normal stimulatory signals) or expressed (e.g. chromosomal amplification may result in overexpression). Proto-oncogenes that are erroneously activated are termed oncogenes.
A number of key tumour suppressor genes, particularly those involved in the regulation of cell cycle control and DNA replication are commonly dysregulated in cancers. However, the frequency of specific gene disruption in tumours arising from different tissue types varies markedly. This holds true for tumours of the urological organs, most notably the frequent loss of the Von Hippel–Lindau tumour suppressor gene (VHL) in renal cancers and activating mutation of the proto-oncogene Fibroblast Growth Factor Receptor in Bladder cancers (see below on “Molecular genetics”).
The Requirements of Tumour Growth: The Hallmarks of Cancer
For cells to persist and proliferate with an increasingly malignant phenotype they must acquire a number of specialized traits that enable them to escape normal cellular control and the growth constraints of their local microenvironment. In their seminal review, cancer biologists Douglas Hanahan and Robert Weinberg [5, 6] identified six key requirements for cancer evolution, these were termed the hallmarks of cancer. The six hallmarks that differentiate tumour cells from the non-malignant cells from which they have originated are common to all cancers, including those of the urological system. However the specific processes and pathways that facilitate the development of the hallmarks vary markedly between specific tumour types. The Hallmarks of cancer, as first outlined by Hanahan and Weinberg [5, 6], are outlined in Table 3.1.
1. Tumour cells must become self sufficient in growth signals |
2. Tumour cells must evade growth suppression |
3. Tumour cells must evade programmed cell death; Apoptosis |
4. Tumour cells are Immortal |
5. Tumour cells induce angiogenesis |
6. Tumour cells are invasive |
1.
Tumour cells must become self sufficient in growth signals. The vast majority of cells in any tissue do not frequently divide; they remain in a replicatively resting state termed quiescence. This is, in part, maintained by a tight control of growth factor molecules (GFs) in the tissue microenvironment. Cancer cells lose the requirement for microenvironment derived GFs through two principal mechanisms; they can acquire the capacity to produce their own GFs (autocrine growth factor stimulation) or/and they acquire mutations that results in cell signaling pathways down-stream of GF stimulation to be permanently switched on [7].
2.
Tumour cells must evade growth suppression. Cell division is tightly regulated. This highly orchestrated process is known as the cell cycle (Fig. 3.2). The initial “decision” to divide occurs at the G1/S phase. Quiescent cells do not proceed through G1 to S phase due to the presence of anti-proliferative signaling molecules secreted by surrounding cells. In common with GFs, these molecules bind trans-membrane receptors that signal to intracellular molecules ensuring the cell does not divide. G1 transition is negatively regulated by a network of tumour suppressor genes and is ultimately controlled by the archetypal tumour suppressor; pRb and its relatives p107 and p130 [8, 9]. Genes involved in the negative regulation of G1/S phase transition are amongst the most frequently mutated in tumours and are common to many cancer types [10] (see section on “Molecular genetics” below).
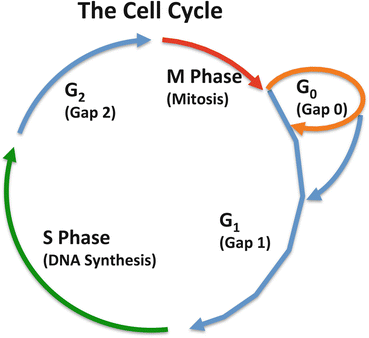
Fig. 3.2
The cell cycle. Differentiated cell in a tissue generally do not replicate, they are quiescent (Gap 0). G0 is maintained by a delicate balance of anti and pro-proliferative signals. Transition from G1 to S-phase is regulated by a complex network of proteins frequently dysregulated in tumour formation. The ultimate signal for a cell to enter S-phase is the Cyclin-dependent kinase mediated inactivation (by phosphorylation) of the retinoblastoma protein (pRB). Once a cell has entered S-phase it is committed to replicate its DNA. Following DNA replication a second checkpoint is reached (G2) where mechanisms ensure that DNA damage has not occurred either by repairing lesions or committing the cell to apoptosis. Release from G2 is followed by mitosis
3.
Tumour cells must evade programmed cell death; Apoptosis. Cancer cells, under the influence of increased mitotic signaling and loss of cell cycle control replicate at an increase rate relative to normal cells. This dysregulated signaling and associated hyperproliferation result in physiological stresses that can trigger signaling pathways that ultimately result in cell death, termed apoptosis. A finely balanced network of pro- and anti- apoptotic proteins determines the fate of cells. Apoptosis is triggered when pro-apoptotic signals outweigh anti-apoptotic signals. A common stress in cancer cells is increased DNA damage, which occurs as a result of hyperproliferation or exposure to mutagens. Elevated levels of DNA damage results in the accumulation of the canonical tumour suppressor p53 [10]. p53 is a transcription factor responsible for elevating the transcription of >100 target genes that direct a variety of cellular responses to stress including pro-apoptotic genes. Inactivation of p53 is the most common genetic defect in tumours; more than half of all tumours have loss of function mutations in p53 [11]. In addition to loss of apoptotic control, inactivation of p53 has wide-ranging pro-oncogenic effects. In tumours that retain wtp53, programmed cell death can be evaded by an imbalance of down-stream pro and anti-apoptotic signaling proteins through genomic alterations.
4.
Tumour cells are Immortal: As discussed above, cells found within a tissue do not generally divide uncontrollably. However, once removed from the constraints of their surrounding microenvironment, either through oncogenic alterations in situ or experimentally removing cells from their constraining microenvironment and growing them in vitro (in tissue culture), cells can grow at a steady rate. However, this growth is not indefinite – there are two well defined barriers to continued cell growth. The first of these barriers is cellular senescence. Senescent cells stop dividing and in tissue culture become enlarged. Although senescent cells no longer have the potential to replicate they are still alive. Ultimately, many of the same intercellular pathways that regulate growth inhibition and aspects of apoptotic control are activated to induce senescence. Through the acquisition of mutations in these pathways senescence can be evaded [12]. Cells that escape senescence continue to divide until a second growth barrier is reached; this barrier, termed crisis, is the result of continued cell replication and is associated with increasing numbers of these cells dying. In vitro, crisis usually results in no viable cells remaining. However, very rarely a cell escapes crisis and become immortal, possessing limitless replicative potential [13].
With every cell division chromosomes become shorter by 50–200 bp, this is a consequence of DNA polymerase not completely replicating the 3’ ends of DNA during division [14]. To ensure that important genetic information is not lost and chromosome end-to end-fusion does not occur, the ends of chromosomes are capped with the repetitive sequence TTAGGG, termed the telomere. Telomeres have an average length of 10–12 kb. Telomere shortening acts a clock measuring cellular replicative age and when a specific telomeric length is reached (4–6 kb) signalling pathways leading to senescence are induced [14]. If senescence is not induced cells may proceed to crisis. For a cell to escape crisis it must be able to maintain telomere length. This is achieved in >90 % of all tumours by the expression of telomerase; an enzyme, generally not expressed in somatic cells, responsible for adding telomere repeat sequences to the end of chromosomes. Expression of telomerase alone is sufficient to immortalize cultured cells [15].
5.
Tumour cells induce angiogenesis: All cells within a given tissue require oxygen and nutrients provided by nearby capillaries. This requirement is such that all cells reside within 100 μM of a capillary. As a tumour forms many of its component cells will find themselves outside of the zone where oxygen and nutrients can readily diffuse, this low level of oxygen (hypoxia) and nutrients is a natural barrier to large tumour formation. To circumvent hypoxia-induced cell death many tumours secrete angiogenic factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) (See “Kidney cancer” below), these factors encourage capillaries to sprout and proliferate towards the tumour [16]. Over-expression of angiogenic factors alone is not sufficient for angiogenesis as many anti-angiogenic factors exist, these are often down regulated in the tumour itself and tumours express high levels of proteinases that degrade anti-angiogenic factors [17]. In combination this results in an imbalance of pro and anti-angiogenic factors that can result in the formation of well vasculated tumours.
6.
Malignant tumours invade the surrounding tissue and metastasis to distant organs: The vast majority of cancer deaths are caused by secondary tumours (metastases) that derive from primary tumour cells invading their local microenvironment followed by entry into the local blood and lymphatic system (intravasation) and eventual invasion into distant organs (extravasation). Once the parenchyma of a new organ has been invaded these micrometastases must acquire the necessary traits to survive and proliferate within the confines of the new microenvironment [18]. This multi-step process requires multiple adaptions and although many of the details are not fully elucidated, it is known that invasive tumour cells have altered expression of cadherins (which determine the adherence of cells to one another) and often overexpress matrix-degrading enzymes. Expression of E-cadherin is required for the correct formation of epithelial tissue sheets and maintaining cell contact induced quiescence [19], and loss of E-cadherin expression is found in many cancers. Conversely, other cadherins, such as N-cadherin, that are involved in cell migration during embryogenesis are often expressed at high levels in aggressively invasive tumour cells [20]. The capacity of a tumour to invade and colonise a particular distant organ is, to some degree dependent upon the site of origin. The most common sites of metastasis for Bladder, Prostate, kidney and testicular cancers are local lymph nodes, bone, lung and liver [21].
Molecular Genetics
Common Molecular Components of Cancers
All tumours have a number of common pathways that are dysregulated, many of these pathways regulate cell cycle progression which is controlled by a number of checkpoints that ensure cells commit to divide only when the appropriate signals are received and once a cell commits to divide it’s DNA is replicated with high fidelity and chromosomal separation occurs correctly prior to cytokinesis [8]. Quiescent cell do not enter the DNA synthesis phase (S phase) of the cell cycle while the canonical tumour suppressor gene product Retinoblastoma protein (pRB) is activated. pRB is a nuclear phosphoprotein that binds to, and inhibits the activity of, the E2F family of transcription factors. Following inactivation of pRB, E2F transcription factors are released and mediate the transactivation of target genes that facilitate the transition from G0/G1 to S phase. These targets include Cyclins, Cyclin dependent kinases, E2F family members themselves, DNA polymerases, DNA repair genes. Also, to ensure control of growth is maintained negative regulators are targeted, these include pRB, p53, p21 and BRCA1 [11]. Loss of functional pRB is common to a number of tumours, particularly retinoblastoma (it was first identified in patients with germline RB1 gene mutations who developed bilateral retinoblastoma), osteosarcoma, lung, melanoma and bladder cancers [22]. The RB1 gene has been found to be mutated in 32 % of sporadic bladder cancers [23]. The presence of RB1 mutations in other urological cancers is low; 7 % of prostate tumours have mutations and less than 1 % of kidney or testicular cancers have RB1 mutations [23].
Loss of cell cycle control is a key requirement for tumour progression and the RB pathway can be considered a central hub of control that is regulated by many tumour suppressors and proto-oncogenes. These controlling proteins themselves often have multiple functions whereby dysregulation results in a contribution to the development of multiple cancer hallmarks.
Following appropriate mitogenic signalling Cyclins D and E and their associated dependent kinases (cyclin dependent kinase 4, 6 and 2) phosphorylate pRB resulting in its inactivation [11]. The cyclins and their dependent kinases are proto-oncogenes frequently found to be over-expressed or constitutively activated through genomic alterations or dysregulation of upstream regulators of their expression. In kidney cancer Cyclin D1 is frequently over-expressed due to loss of the tumour suppressor pVHL, which influences many cancer-related pathways and networks. The activity of Cyclin/CDKs is a negatively regulated by a family of proteins called CDK Inhibitors. These include p16INK4a, p15INK4b, p18 INK4c and p19INK4d that inhibit the D type cyclin dependent kinases 4 and 6 and the CIP/KIP family, whose members (p21CIP1/WAF1, p27KIP1, p57KIP2) inhibit the activity of all CDKs, these proteins are commonly under-expressed or lost in multiple tumour types [24]. Loss of p16INK4a is a common, early event in the evolution of bladder cancers [25]. Following DNA damage, or other physiological stresses, p53 is activated by phosphorylation resulting in the up-regulation of cell cycle inhibitory genes such as p21CIP1/WAF1 or pro-apoptotic genes such as PUMA and BAX [26]. p53 is the most commonly dysregulated gene in tumours, it may be lost by deletion or epigenetic dysregulation or by point mutation, including dominant negative mutations that cause p53 to form inactive tetramers [27].
Genetics of Genito-Urinary Cancers
Kidney Cancer
The most common type of kidney cancer is Renal Cell Carcinoma (RCC). RCC is derived from proximal tubular epithelial cells. The majority of RCC (~75 %) are classified as clear cell (conventional), the next most common type are papillary tumours (~15 %) [28].
Unlike many other tumours, in RCC the tumour suppressor genes TP53 and RB1 are infrequently deactivated by direct mutation (TP53 is mutated in 11 % and RB1 in <1 % of all kidney cancers, while CDKN2A-p16 is mutated in 10 %) [23]. The low frequency of mutation in these key tumour suppressors indicate a significant difference in the pathogenesis of RCC compared to many other neoplasms. Deletion of the short arm of chromosome 3 is the most common genetic aberration in RCC occurring in up to 90 % of clear cell RCC. However 3p loss is not associated with papillary RCC [29].
VHL
The VHL gene was identified by studying patients with the Von Hippel-Lindau disease, familial kidney cancer syndrome, which is caused by germline VHL gene mutations [30]. The VHL gene maps to the short arm of chromosome 3 (3p25.3) and the most frequent genetic event in the evolution of clear cell RCC is the Inactivation of VHL. The VHL protein binds to elongin B, elongin C and cullin 2 to form a ubiquitin ligase complex [31] that facilitates the ubiquitination of target proteins leading to their proteosomal degradation. The best studied of these targets are the Hypoxia Inducible Factors HIF1-α and HIF2-α subunits (form herein referred to as HIF-α) that in combination with HIF-β form the Hypoxia Inducible Factor HIF, a transcription factor [32]. Under normal oxygen conditions (normoxia) HIF-α is targeted for VHL-directed ubiquitination by the hydroxylation of a crucial proline residue by the enzyme HIF Prolyl Hydroxylase (PHD) [33]. Hence, in the presence of functioning (wild-type) VHL, and under normoxic conditions, HIF is rapidly degraded. However loss of both copies (alleles) of VHL results in the accumulation of HIF under normoxic conditions and elevated levels of its target genes [32].
The Target genes of HIF are regulatory units of several key pathways that can contribute to cancer hallmarks, notably angiogenesis by the upregulation of: Vascular Endothelial Growth Factor (VEGF), Platelet Derived Growth Factor β (PDGFβ), Transforming Growth Factor β (TGFβ) and Connective Tissue Growth Factor (CTGF), Invasion and metastasis by the up-regulation of: Stromal cell derived Factor (SDF) CXC chemokine receptor 4 (CXCR4), Matrix Metallopeptidase 1 (MMP1) and c-MET, Proliferation and Survival by the up regulation of: Cyclin D1 and Transforming Growth factor α (TGF α). Cell metabolism is also up regulated by HIF dependent increased expression of genes involved in glucose uptake and metabolism: Hexokinase 2 (HK2) and pyruvate dehydrogenase kinase (PDK1) and genes involved in acid/base regulation: Carbonic dehydrogenase 9 (CA IX) and Carbonic dehydrogenase 12 (CAXII) [34]. Clear cell RCC is a highly angiogenic tumour type, resulting from the dysregulation of multiple positive regulators of angiogenesis following the loss of a single tumour suppressor gene (VHL). In addition to regulating HIF1 levels, VHL has multiple HIF-independent functions but it is unclear how they may contribute to RCC. Although Loss of VHL is extremely common in clear cell RCC it is not sufficient alone for tumorigenesis to occur, further oncogenic events are required.
PBRM1
In 2011, following a screen of clear cell RCC by whole exome sequencing truncating mutations were found in the PBRM1 gene which maps to chromosome 3p21 and encodes BAF 180 (Polybromo 1) a component of the pBAF SWI/SNFB chromatin re-modelling complex [35]. Chromatin re-modelling associated with pBAF SWI/SNFB is implicated in replication, transcription, DNA repair and control of proliferation and differentiation. PBRM1 truncating mutations were found in 41 % of clear cell RCC, Many of these tumours had also lost functional VHL. Three other genes involved in histone modification have been identified as mutated (at a lower frequency than PBRM1), these are SETD2 or JARID1C and UTX [36] suggesting that, in addition to VHL related gene dysregulation, modifying gene expression by an epigenetic mechanism plays a principal role in the formation of many renal tumours.
c-MET
c-MET is a cell surface transmembrane receptor for hepatocyte growth factor (HGF). Activated c-MET signals, via RAS and PI3K, to the Mitogen-Activated Protein Kinase (MAPK) cascade resulting in proliferation and (upon sustained activation) Invasion [37]. Activating mutations of the tyrosine kinase domain of c-MET have been found in 13 % of sporadic papillary RCC [38]. Moreover, amplifications of the region of chromosome 7 where the cMET gene lies and chromosome 7 trisomy are common in papillary RCC [39]. Up-regulation of cMET can result in receptor auto-activation that, as with activating mutations, negates the requirement for HGF. PTEN, a negative regulator of the MET-PI3K pathway has been found to be mutated in approximately 3 % of RCC (COSMIC) and Hepatocyte growth factor activator Inhibitor 2 (HAI-2) which prevents activation of c-MET by HGF is frequently down regulated or silenced by epigenetic mechanisms [40]. Activating mutations of the RAS family of proto-oncogenes is rare in RCC.
RASSF1
RASSF1 encodes a protein that regulates cell cycle at the G1/S phase and G2/M phase transitions and also induce apoptosis, it is frequently epigenetically inactivated in RCC by promoter hypermethylation [41].
Bladder Cancer
The vast majority of Bladder cancers (90 %) arise from the urothelial cells that line the bladder (Urothelial carcinoma; UCa or Transitional Cell Carcinoma; TCC). These cancers are traditionally classified by their histopathology and are either non-muscle invasive bladder cancer (NMIBC) or muscle invasive bladder cancer (MIBC) bladder cancer that grow into the muscle layer of the bladder. The remaining 10 % of tumours are either squamous cell, derived from the bladder lining cells (8 %) or adenocarcinoma, derived from the mucous producing cells (1–2 %) [42].
Loss of the short arm of chromosome 9 (9p) is common to both nonmuscle and muscle invasive bladder cancers. The tumour suppressor CDKN2A (p16/ARF) is present in this region. Chromosome deletion, point mutation and promoter methylation resulting in complete loss of p16/ARF activity is common [43, 44]. Other Tumour suppressor genes present on 9p include the interferon-α (INF–α) gene, the product of which is involved in apoptotic control [45], and Tuberous Sclerosis 1 (TSC1) the product of which is involved in the regulation of multiple processes including cellular growth [46].
Loss of Chromosome 9p is an early event in bladder cancer that results in the proliferation of a pre-neoplastic clone that is phenotypically similar to normal urothelium. The proliferation of a large number of phenotypically ‘normal’ cells with an increased neoplastic potential is known as a field effect because there is a large number of cells (the field) from which additional genomic alterations may occur in a single cell for tumorigenic clonal expansion to occur [47].
Two separate genetic mechanisms lead to the development of the two main phenotypic variants of bladder cancers, both arise from a pre-neoplastic field. 70–80 % of TCC are low-grade non-invasive papillary tumours, these commonly have activating mutations in the Fibroblast Growth Factor receptor 3 gene (FRGF3), this is the most commonly mutated gene in bladder cancers (45 %) [23]; over-expression of FGFR3 is also common [48]. Fibroblast Growth Factor receptor 3 is a tyrosine kinase receptor that (in a similar manner to c-MET (see “Kidney cancer” below)), when activated, signals to the MAPK cascade via RAS and PI3K [49]. Activating mutations of the RAS proto-oncogene are also common (9 %;) [23] as are activating mutations of PIK3CA (20 %;) [23], the gene that encodes the catalytic subunit of PI3K.
Loss of p53 and/ or pRB activity in cells found within a pre-neoplastic field is associated with the development of Carcinoma In Situ (CIS) that frequently develops into high-grade invasive UCa. These highly invasive and metastatic tumours often over-express the angiogenic factor VEGF, matrix metalloproteinases (MMPs) and have reduced expression of the epithelial cell adhesion molecule E-cadherin. Approximately 15 % of low-grade non-invasive papillary tumours progress to high-grade invasive tumours, this progression is associated with dysregulation of p53 and/or pRB and loss of chromosomes 8p, 11p and 13q which harbour the known tumour suppressor genes SFRP1, WT1, and RB1 respectively [48]. As is common in RCC the tumour suppressor gene RASSF1 is frequently silenced by promoter methylation in bladder cancer [50].
Prostate Cancer
Prostate cancer can be regarded as a multifocal disease as primary tumours often contain histologically and genetically distinct regions. However, the genomes of cells in advanced prostate cancer are similar indicating that these advanced tumours have undergone clonal evolution. The majority of prostate tumours are adenocarcinomas derived from the epithelium. Prostatic Intraepithelial Neoplasia (PIN), which is characterized by luminal epithelial hyperplasia and a reduction of basal cells. PIN cells often have variation in the size and shape of their nuclei consistent with increased DNA content [51, 52].
Of the known Cancer-related genes, p53 is the most commonly mutated (19 %: COSMIC) in prostate cancer. The MYC oncogene is upregulated in many PIN lesions, However, this up-regulation is often not associated with chromosomal amplification (8q24). Genetic studies to identify inherited factors that predispose to prostate cancer found that genetic variants at chromosomal region 8q24 predisposed to prostate cancer by disturbing regulation of the MYC proto-oncogene [53]. The MYC containing 8q24 region is amplified in a significant sub-set of advanced metastatic prostate cancers [54]. Inactivation of the PTEN tumour suppressor gene (an inhibitor of the AKT/mTOR signalling pathway) is a common early event in prostate carcinogenesis [55] and inactivating mutations are also observed in 14 % of tumours (COSMIC). Less frequently the AKT/mTOR pathway is upregulated by activating mutations of AKT and PI3K (COSMIC). The RAS/RAF MAPK pathway is frequently upregulated through a paracrine mechanism in advanced prostate cancers [56] and less commonly via activating mutations of RAS (5 %: COSMIC) or RAF (4 %: COSMIC).
NKX3.1
NKX3.1 is a homeobox gene that codes for the homeodomain-containing transcription factor NKX3-1. NKX3-1 is predominantly expressed in the prostate and in an androgen dependent manner. NKX3.1 is found within a 150 Mb region of Chromosome 8q21 that displays loss of heterozygosity (LOH, a marker of chromosomal alterations in cancer) in up to 85 % of high grade PIN lesions and adenocarcinomas, the frequency of LOH increases with increased grade of tumour [57]. Reduced expression of NKX3.1 is associated with a reduced response to oxidative DNA damage [58]. It is thought that NKX3.1 may act as a haploinsufficient tumour suppressor and that inactivation may be a key initiating event in the evolution of prostate cancer.
Androgen Receptor (AR)
AR is a member of the steroid hormone receptor family of ligand-activated nuclear transcription factors, its principal ligand is testosterone. AR plays a key role in normal prostate development and prostate cancer. Following androgen binding, AR is translocated to the nucleus where it acts as a transcription factor for a wide range of target genes via binding to hormone response elements in their promoters [59]. In relation to tumour formation, activation of AR positively regulates genes involved in cell proliferation and anti-apoptotic signaling [60].
Intact AR signaling is necessary for the development of prostate cancer; AR is present in primary and metastatic prostate cancers irrespective of tumour stage. Treatment for advanced prostate cancer is often surgical or chemical castration to reduce the level of testosterone signalling to AR. This treatment leads to rapid tumour apoptosis however relapse is common. In approximately 30 % of castration resistant tumours the AR gene copy number is amplified [61] a further 10–30 % acquire gain of function mutations that increase AR activity via different mechanisms including increased sensitivity to androgens, the capacity to respond to other hormones, auto-activation or increased protein stability [62]. In addition, metastatic tumours can express enzymes that synthesize androgens or convert other hormones into testosterone [63].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


