Lisa M. Guay-Woodford
Other Cystic Kidney Diseases
In addition to autosomal dominant polycystic kidney disease (ADPKD), numerous other disorders share renal cysts as a common feature1,2 (Table 47-1). These disorders may be inherited or acquired; their manifestations may be confined to the kidney or expressed systemically. They may present at a wide range of ages, from the perinatal period to old age (Fig. 47-1). The renal cysts may be single or multiple, and the associated renal morbidity may range from clinical insignificance to progressive parenchymal destruction with resultant renal impairment.
Table 47-1
Renal cystic disorders.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM.
Renal Cystic Disorders
Nongenetic
Developmental
Medullary sponge kidney*
Renal cystic dysplasia
Multicystic dysplasia
Cystic dysplasia associated with lower urinary tract obstruction
Diffuse cystic dysplasia: syndromal and nonsyndromal
Acquired
Simple cysts
Solitary multilocular cysts
Hypokalemic cystic disease
Acquired cystic disease (in advanced renal failure)
Genetic
Autosomal Dominant
Autosomal dominant polycystic kidney disease
Adult-onset medullary cystic disease
Tuberous sclerosis
von Hippel–Lindau disease/syndrome
Autosomal Recessive
Autosomal recessive polycystic kidney disease
Juvenile nephronophthisis
Meckel-Gruber syndrome
X-Linked
Orofaciodigital syndrome type I
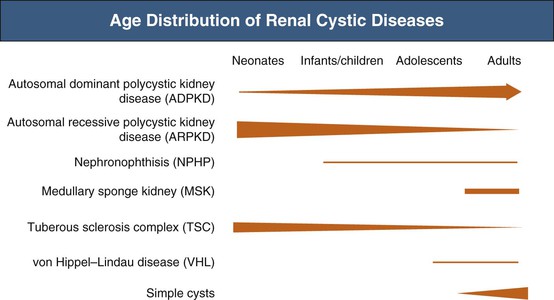
The clinical context often helps distinguish these renal cystic disorders from one another. Echogenic, enlarged kidneys in a neonate or infant should suggest autosomal recessive polycystic kidney disease (ARPKD), ADPKD, tuberous sclerosis complex (TSC), or one of the many congenital syndromes associated with renal cystic disease. Renal impairment in an adolescent suggests juvenile nephronophthisis–medullary cystic disease complex or ARPKD as possible etiologies. The finding of a solitary cyst in a 5-year-old child may indicate a calyceal diverticulum, whereas this finding in a 50-year-old patient is most compatible with a simple renal cyst. Renal stones occur in ADPKD and medullary sponge kidneys. For disorders with systemic manifestations, such as ADPKD, TSC, and von Hippel–Lindau disease, the associated extrarenal features may provide other important differential diagnostic clues.
For an increasing number of the single-gene disorders, genetic testing is available in expert laboratories around the world. Genetic testing resources are listed at Gene Tests (http://www.genetests.org) and the U.S. National Institutes of Health (NIH) Genetic Testing Registry (http://www.ncbi.nlm.nih.gov/gtr).
Autosomal Recessive Polycystic Kidney Disease
Definition
Autosomal recessive polycystic kidney disease (ARPKD) is a severe, typically early-onset form of cystic disease that primarily involves the kidneys and biliary tract. Affected patients have a spectrum of clinical phenotypes that depend in part on the age at presentation.3
Etiology and Pathogenesis
Genetic Basis of ARPKD
All typical forms of ARPKD are caused by mutations in a single gene, PKHD1 (polycystic kidney and hepatic disease), which encodes multiple, alternatively spliced isoforms predicted to form both membrane-bound and secreted proteins.4 The largest protein product of PKHD1, the fibrocystin/polyductin complex (FPC), contains one membrane-spanning domain and an intracellular C-terminal tail. FPC localizes, at least in part, to the primary cilium and the centrosome in renal epithelial cells.5 The basic defects observed in ARPKD suggest that FPC mediates the terminal differentiation of the collecting duct and biliary tract. However, the exact function of the numerous isoforms has not been defined, and the widely varying clinical spectrum of ARPKD may depend partly on the nature and number of splice variants that are disrupted by specific PKHD1 mutations.
Pathogenetic Mechanisms
ARPKD typically begins in utero, and the renal cystic lesion appears to be superimposed on a normal developmental sequence. The tubular abnormality primarily involves fusiform and/or saccular dilation of the collecting ducts. Microdissection studies have excluded tubular obstruction as a primary pathogenic mechanism. The biliary lesion involves defective remodeling of the ductal plate in utero. As a result, primitive bile duct configurations persist, and progressive portal fibrosis evolves.6 The remainder of the liver parenchyma develops normally. The defect in ductal plate remodeling is accompanied by abnormalities in the branching of the portal vein. The resulting histopathologic pattern is termed congenital hepatic fibrosis.
Autosomal recessive PKD is one of a number of renal cystic diseases associated with congenital hepatic fibrosis, prompting these disorders to be described as hepatorenal fibrocystic diseases.7 The primary cilium appears to play a central role in the pathogenesis ARPKD and other hepatorenal fibrocystic diseases,8 and thus this set of disorders is encompassed under the broader term ciliopathies, in which dysfunction of the cilia-centrosome complex appears to underpin the development of a wide array of phenotypes including renal cystic disease.9
Epidemiology
The estimated incidence of ARPKD is 1 per 20,000 live births. It occurs more frequently in Caucasians than in other ethnic populations.3
Clinical Manifestations
The clinical spectrum of ARPKD is variable. The majority of cases are identified either in utero or at birth. The most severely affected fetuses have enlarged echogenic kidneys and oligohydramnios caused by poor fetal urine output. These fetuses develop the “Potter” phenotype, with pulmonary hypoplasia, a characteristic facies, and deformities of the spine and limbs. At birth, these neonates often have a critical degree of pulmonary hypoplasia that is incompatible with survival. The estimated perinatal mortality is about 30%. Although frequently compromised, renal function is rarely a cause of neonatal death.
For those who survive the first month of life, the reported mean 5-year patient survival is 85% to 90%.3,10 Morbidity and mortality results from severe systemic hypertension, renal impairment, and portal hypertension from portal-tract hyperplasia and fibrosis.3,10,11 Hypertension usually develops in the first several months and ultimately affects 70% to 80% of patients. ARPKD patients have defects in both urinary diluting capacity and concentrating capacity. Newborns can have hyponatremia, presumably resulting from defects in free water excretion.3 Net acid excretion may be reduced, but metabolic acidosis is not a significant clinical feature.12 Retrospective studies have noted an increased incidence of pyuria on urinalysis, as well as culture-confirmed urinary tract infections (UTIs).3
In the first 6 months of life, ARPKD infants may have a transient improvement in glomerular filtration rate (GFR) caused by renal maturation. Subsequently, a progressive but variable decline in renal function occurs, with patients presenting in the first month of life progressing more rapidly to end-stage renal disease (ESRD) than those presenting at more than one month of age.12 With advances in effective therapy for ESRD, prolonged survival is common, and for many patients the hepatic complications become the predominant clinical issue.
On average, infants with serum creatinine values above 2.2 mg/dl (200 µmol/l) progress to ESRD within 5 years, but this is highly variable. In longitudinal studies, the probability of renal survival without ESRD is about 85% at 1 year, 70% at 10 years, 65% at 15 years, and 40% at 20 years.10,11
In children who present later in childhood or in adolescence, portal hypertension is frequently the predominant clinical abnormality, with hepatosplenomegaly and bleeding esophageal or gastric varices, as well as hypersplenism with consequent thrombocytopenia, anemia, and leukopenia. Hepatocellular function is usually preserved. Ascending suppurative cholangitis is a serious complication and can cause fulminant hepatic failure.13,14
Other associated features include growth restriction,3 although the mechanism is not yet defined, and rarely intracranial aneurysms.15
Pathology
Kidney
The renal involvement is invariably bilateral and largely symmetric. The histopathology varies depending on the age of presentation and the extent of cystic involvement (Fig. 47-2, A and B).
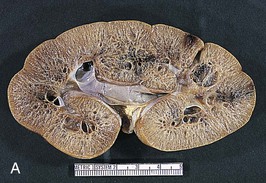
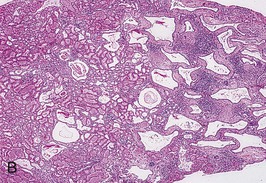
In the affected neonate, the kidneys can be 10 times normal size but can retain the typical kidney contour. Dilated, fusiform collecting ducts extend radially through the cortex. In the medulla, the dilated collecting ducts are more often cut tangentially or transversely. Up to 90% of the collecting ducts are involved. Associated interstitial fibrosis is minimal in neonates and infants, but increases with progressive disease.
In patients diagnosed later in childhood, the kidney size and extent of cystic involvement tend to be more limited. Cysts can expand up to 2 cm in diameter and assume a more spherical configuration. Progressive interstitial fibrosis is probably responsible for secondary tubular obstruction. In older children, medullary ductal ectasia is the predominant finding.
Cysts are lined with a single layer of nondescript cuboidal epithelium. The glomeruli and nephron segments proximal to the collecting ducts are initially structurally normal, but are often crowded between ectatic collecting ducts or displaced into subcapsular wedges. The presence of cartilage or other dysplastic elements indicates a diagnosis other than ARPKD, such as cystic dysplasia.
Liver
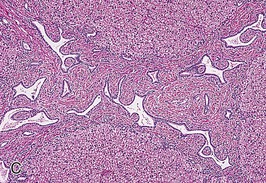
The liver lesion in ARPKD is characterized by ductal plate malformation.6 The liver can be either normal in size or somewhat enlarged. Bile ducts are dilated (biliary ectasia) and marked cystic dilation of the entire intrahepatic biliary system (Caroli syndrome) is well described.16 In neonatal ARPKD, the bile ducts are increased in number, tortuous in configuration, and often located around the periphery of the portal tract. In older children, the biliary ectasia is accompanied by increasing portal tract fibrosis and hypoplasia of the small portal vein branches (Fig. 47-2, C). The hepatic parenchyma may be intersected by delicate fibrous septa that link the portal tracts, but the hepatocytes themselves are seldom affected.
Diagnosis
Autosomal recessive PKD must be differentiated from a range of other pediatric renal cystic disorders (Table 47-2).
Table 47-2
Features of pediatric renal cystic disease.
ARPKD, Autosomal recessive polycystic kidney disease; CM, corticomedullary.
| Features of Pediatric Renal Cystic Disease | ||||||
| Feature | ARPKD | NPHP | Meckel-Gruber1 | GCKD2 | ADPKD | TSC |
| Clinical Characteristics | ||||||
| Clinical onset | Perinatal | NPHP2: 0-5 yr NPHP: 10-18 yr | Perinatal, infancy | Infancy, older children | Infancy,3 older children | Infancy,3 older children |
| Enlarged kidneys | Yes | NPHP2: yes NPHP3: some cases NPHP: no | Yes | No | Occurs | Occurs |
| Renal pathology | Multiple cysts | NPHP2: multiple cysts NPHP: few cysts at CM junction | Multiple cysts | Multiple cortical cysts | Multiple cysts | Few to multiple cysts; angiomyolipoma |
| Cyst infection | Uncommon | No | Uncommon | No | Occurs | Uncommon |
| Blood pressure | Normal/increased | NPHP2: increased NPHP: normal | Normal | Normal/ increased | Normal/ increased | Normal/increased |
| Renal function | Normal/impaired | Normal/impaired | Normal/impaired | Normal | Normal | Normal |
| Nephrocalcinosis/nephrolithiasis | Nephrocalcinosis up to 25% | No | No | No | Nephrolithiases occur | No |
| CHF4 | Yes | Rare | Yes | No | 10%-15% infantile ADPKD | No |
| Pancreas lesions | No | No | No | MODY5 | No | No |
| CNS involvement | No | (Joubert)5 | Encephalocele; mental retardation | No | No | Seizures, mental retardation |
| Genetics | ||||||
| Disease gene | PKHD1 | NPHP1-NPHP15 | MSK1-MSK10 | PKD1 TCF2 | PKD1 PKD2 | TSC1 TSC2 |
| Genetic testing6 | Yes | Most* | Most* | Yes | Yes | Yes |
1 Meckel-Gruber syndrome (MKS) is a severe, often lethal autosomal recessive disorder characterized by bilateral renal cystic dysplasia, biliary ductal dysgenesis, bilateral postaxial polydactyly, and variable central nervous system (CNS) malformations. The triad of renal cystic disease, occipital encephalocele, and polydactyly is most common. Genes disrupted in MKS have also been identified in nephronophthisis (NPHP) and Joubert patients, suggesting a phenotypic spectrum.
2 Glomerulocystic kidney disease (GCKD) can occur as the infantile manifestation of autosomal dominant polycystic kidney disease (ADPKD). Familial hypoplastic GCKD, caused by mutations in TCF2, the gene encoding hepatocyte nuclear factor (HNF-1β), can be associated with maturity-onset diabetes of the young, type 5 (MODY5).
3 A contiguous germ-line deletion of both the PKD1 and the TSC2 gene (the PKDTS contiguous gene syndrome; MIM 600273) occurs in a small group of patients with features of tuberous sclerosis complex (TSC), as well as massive renal cystic disease reminiscent of ADPKD, severe hypertension, and a progressive decline in renal function with the onset of end-stage renal disease (ESRD) in the second or third decade of life.
4 Congenital hepatic fibrosis (CHF).
5 Joubert syndrome (JBTS; MIM 213300) is a genetically heterogeneous (JBTS 1-20), autosomal recessive disorder characterized by developmental defects in the cerebellum (cerebellar vermis aplasia) and the eye (coloboma), as well as retinitis pigmentosa, congenital hypotonia, and either ocular motor apraxia or irregularities in breathing patterns during the neonatal period. The disease can be associated with NPHP, and mutations in several NPHP genes have been described in JBTS patients.
6 Genetic testing: listed at GeneTests (http://www.genetests.org) and the NIH Genetic Testing Registry (http://www.ncbi.nlm.nih.gov/gtr).
* Testing is available for most NPHP and MKS genes.
Imaging
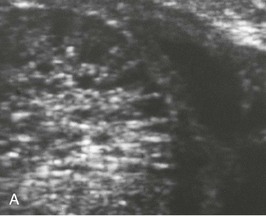
In the past decade, clinical diagnosis has increasingly relied on imaging instead of histopathologic analysis. ARPKD belongs to the hepatorenal fibrocystic diseases,7 most of which are characterized by large, echogenic kidneys in the fetus and neonate. However, recent studies indicate that to a large extent these disorders can be distinguished by ultrasound.17 ARPKD kidneys in utero are hyperechogenic and display decreased corticomedullary differentiation because of the hyperechogenic medulla (Fig. 47-3, A). With high-resolution ultrasound, the radial array of dilated collecting ducts may be imaged. In comparison, ADPKD kidneys in utero tend to be moderately enlarged with a hyperechogenic cortex and relatively hypoechogenic medulla causing increased corticomedullary differentiation.
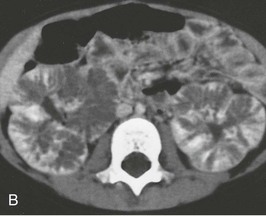
Kidney size typically peaks at 1 to 2 years of age, then gradually declines relative to the child’s body size, and stabilizes by 4 to 5 years. As patients age, there is increased medullary echogenicity, with scattered small cysts measuring less than 2 cm in diameter. These cysts and progressive fibrosis can alter the usual kidney contour, causing ARPKD in some older children to be mistaken for ADPKD. Contrast-enhanced computed tomography (CT) scanning can be useful in delineating the renal architecture in these children (Fig. 47-3, B). Bilateral pelvicaliectasis and renal calcifications have been reported in 25% and 50% of ARPKD patients, respectively.11,18 In adults with medullary ectasia alone, the cystic lesion may be confused with medullary sponge kidney.
The liver may be either normal in size or enlarged. It is usually less echogenic than the kidneys. Prominent intrahepatic bile duct dilation suggests associated Caroli syndrome. With age, the portal fibrosis tends to progress, and in older children, ultrasound typically shows hepatosplenomegaly and a patchy increase in hepatic echogenicity.16,19
Genetic Testing
With the identification of PKHD1 as the principal disease gene in ARPKD, genetic testing is available as a clinical diagnostic tool. The mutation detection rate is 80% to 87%. Current diagnostic algorithms include gene-based analysis and haplotype-based genotyping (in informative families).20,21 Genetic testing is primarily applied in the context of prenatal testing and preimplantation genetic diagnosis.22 To date, there is limited evidence for genotype-phenotype correlations, except perhaps in affected fetuses.23 Although patients with two truncating mutations have been reported to have a higher risk of perinatal demise,21,24 recent studies identified a large, homozygous deletion in an 8-year-old boy.25 ARPKD is associated with a high frequency of unique, missense changes in PKHD1, which can complicate the unequivocal interpretation of gene-based testing. Moreover, about 20% of ARPKD siblings have discordant clinical phenotypes.10 These data may complicate genetic counseling, and caution must be exercised in predicting the clinical outcome of future affected children.26
Treatment
The survival of ARPKD neonates has improved significantly in the last two decades because of advances in mechanical ventilation for neonates and other supportive measures. Aggressive interventions such as unilateral or bilateral nephrectomies and continuous hemofiltration have been advocated in neonatal management, but prospective, controlled studies have yet to be performed.
For those children who survive the perinatal period, blood pressure control is a major clinical issue. Angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), adrenergic antagonists, and loop diuretics are effective antihypertensive agents. The management of ARPKD children with declining GFR should follow the standard guidelines established for chronic kidney disease (CKD) in pediatric patients.27
Given the relative urinary concentrating defect, ARPKD children should be monitored for dehydration during intercurrent illnesses associated with fever, tachypnea, nausea, vomiting, or diarrhea. For infants with severe polyuria, thiazide diuretics may be used to decrease distal nephron solute and water delivery. Acid-base balance should be closely monitored and supplemental bicarbonate therapy initiated as needed.
Close monitoring for portal hypertension is warranted in all ARPKD patients. There is no correlation between the severity of the kidney and liver disease.3,19 Recent studies suggest that the platelet count combined with serial abdominal ultrasound (assessing liver and splenic size) and Doppler flow studies provide good surrogate markers for the portal hypertension severity.19 Medical management may include sclerotherapy or variceal banding.16 Surgical approaches such as portocaval or splenorenal shunting may be indicated in some patients. Although hypersplenism occurs quite often, splenectomy is seldom warranted. Unexplained fever with or without elevated transaminase levels suggests bacterial cholangitis and requires meticulous evaluation, sometimes including a percutaneous liver biopsy, to make the diagnosis and guide aggressive antibiotic therapy.
Effective management of systemic and portal hypertension, coupled with successful renal replacement therapy, has allowed long-term patient survival. Therefore, the prognosis in ARPKD, particularly for children who survive the first month of life, is much less bleak than generally thought, and aggressive medical therapy is warranted.
Current treatment of patients with ARPKD is entirely supportive, although preclinical studies suggest that new, targeted therapies may have future benefit for this cohort of patients.28
Transplantation
A prolonged period of dialysis in childhood has been associated with both cognitive and educational impairment. Therefore, renal transplantation is the treatment of choice for ESRD in ARPKD patients, and at least one report advocates preemptive nephrectomy in neonates with greatly enlarged kidneys.29 ARPKD is a recessive disorder, and therefore either parent may be a suitable kidney donor. However, the recent identification of subtle renal and liver ultrasound abnormalities in ARPKD parents30 suggests caution that requires more extensive follow-up analyses. Native nephrectomies may be warranted in patients with massively enlarged kidneys to allow allograft placement.
In some patients, combined kidney-liver transplantation may be appropriate.31 Indications include the combination of renal failure and either recurrent cholangitis or significant complications of portal hypertension (e.g., recurrent variceal bleeding, refractory ascites, hepatopulmonary syndrome). In addition, liver transplantation may be a consideration for patients with a single episode of cholangitis in the context of marked abnormalities in the biliary system (Caroli syndrome).16
Juvenile Nephronophthisis–Medullary Cystic Disease Complex
Definitions
Juvenile nephronophthisis (NPHP) and medullary cystic kidney disease (MCKD) share the same triad of histopathologic features: tubular basement membrane irregularities, tubular atrophy with cyst formation, and interstitial cell infiltration with fibrosis. These histopathologically similar disorders differ only in their mode of transmission, age of onset, and genetic defects. NPHP is an autosomal recessive disorder that presents in childhood, whereas MCKD is an autosomal dominant disorder that occurs in adults. The inclusive term juvenile nephronophthisis–medullary cystic kidney disease complex has been used to describe these disorders. NPHP is much more common than MCKD, however, and NPHP has been reported both as an isolated renal disease and in association with a wide range of systemic manifestations, including retinitis pigmentosa, congenital hepatic fibrosis, oculomotor apraxia, and skeletal anomalies. Therefore, these entities are considered separately.
Autosomal Recessive Juvenile Nephronophthisis
An autosomal recessive tubulointerstitial nephropathy, NPHP is one of the most frequent inherited causes of ESRD in children and adolescents.32 The term nephronophthisis derives from the Greek, meaning “progressive loss of nephrons.”
Genetic Basis of NPHP
Multiple disease-causing genes have been identified in NPHP patients. Defects in NPHP1 account for 21% of NPHP, with large, homozygous deletions detected in 80% of affected family members and in 65% of sporadic cases. Mutations in each of the remaining NPHP genes cause no more than 3% of NPHP-related disease.32 Clinical disease expression seems to be exacerbated by oligogenic inheritance, that is, patients carrying two mutations in a single NPHP gene, as well as a single-copy mutation in an additional NPHP gene. In addition, multiple allelism, or distinct mutations in a single gene, appears to explain the continuum of multiorgan phenotypic abnormalities observed in NPHP, Meckel syndrome, and Joubert syndrome.33,34
Most of the protein products of the NPHP-associated genes are expressed in the cilia-centrosome complex,32,34 and NPHP is considered as one of the ciliopathies.
Clinical Manifestations
Renal Disease
Three distinct forms of NPHP—infantile, juvenile, and adolescent—were initially described, based on the age of onset of ESRD. In the infantile form, ESRD consistently occurs before 5 years of age, whereas in juvenile NPHP, the most common form, ESRD occurs at a mean age of 13 years. However, no clear genotype-phenotype correlation exists for this spectrum of presentations, and these disorders should be referred to with the single designation, NPHP.35,36
Decreased urinary concentrating capacity is invariable in NPHP and usually precedes the decline in renal function, with typical onset between 4 and 6 years of age. Polyuria and polydipsia are common. Salt wasting develops in most patients with renal impairment, and sodium supplementation is often required until the onset of ESRD. One third of patients become anemic before onset of renal impairment, attributed to a defect in the functional regulation of erythropoietin production by peritubular fibroblasts.37 Growth restriction disproportionate to the degree of renal impairment is a common finding.
Slowly progressive decline in renal function is typical of NPHP. Although symptoms can be detected after age 2 years, they may progress insidiously, such that 15% of affected patients are recognized only after ESRD has developed. There is no specific treatment. The disease is not known to recur in renal allografts.
Children with the infantile variant develop symptoms in the first few months of life and rapidly progress to ESRD, usually before age 2 years but invariably by age 5. Severe hypertension is common in this disorder.
Unlike patients with polycystic kidney disease or medullary sponge kidney, NPHP patients rarely develop flank pain, hematuria, hypertension, UTIs, or renal calculi.
Associated Extrarenal Abnormalities
Extrarenal abnormalities have been described in approximately 10% to 15% of NPHP patients.38 The most frequently associated anomaly is retinal dystrophy caused by tapetoretinal degeneration (Senior-Loken syndrome). Severely affected patients present with coarse nystagmus, early blindness, and a flat electroretinogram (Leber congenital amaurosis); whereas those with moderate retinal dystrophy typically have mild visual impairment and retinitis pigmentosa. Other extrarenal anomalies include oculomotor apraxia (Cogan syndrome), cerebellar vermis aplasia (Joubert syndrome), and cone-shaped epiphyses of the bones. Congenital hepatic fibrosis occurs on occasion in NPHP patients, but the associated bile duct proliferation is mild and qualitatively different from that in ARPKD.
Pathology
In classic NPHP, the kidneys are moderately contracted, with parenchymal atrophy causing a loss of corticomedullary demarcation. Histopathological findings include tubular atrophy with thickened tubular basement membrane, diffuse and severe interstitial fibrosis, and cysts of variable size distributed in an irregular pattern at the corticomedullary (CM) junction and in the outer medulla. However, up to 25% of NPHP kidneys have no grossly visible cysts.
In the typical NPHP renal lesion, clusters of atrophic tubules either alternate with groups of viable tubules showing dilation or with marked compensatory hypertrophy or with groups of collapsed tubules. Multilayered thickening of tubular basement membranes is a prominent histopathologic feature (Fig. 47-4). This pattern is not unique, but the abrupt transition from one type of tubular profile to another suggests NPHP. Moderate interstitial fibrosis, usually without a significant inflammatory cell infiltrate, is interspersed among the atrophic tubules. Spherical, thin-walled cysts lined with a simple cuboidal epithelium may be evident at the CM junction, in the medulla, and even in the papillae. Microdissection studies indicate that these cysts arise from the loop of Henle, distal convoluted tubules, and collecting ducts. Glomeruli may be normal, although some may be completely sclerosed. Other glomeruli show periglomerular fibrosis, and still others have dilation of Bowman space, suggestive of glomerulocystic kidney disease.39
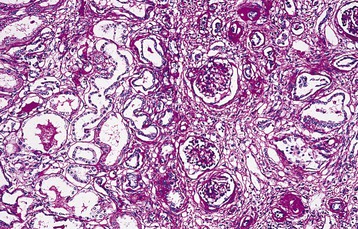
In comparison, the infantile form has features of both classic NPHP, such as tubular cell atrophy, interstitial fibrosis, and tubular cysts, and polycystic kidney disease (PKD), including enlarged kidneys and widespread cystic involvement.36
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


