Walter H. Hörl † Normal hemostasis begins with platelet adhesion to vascular endothelium and requires a relatively vasoconstricted vessel wall, integrity of platelet glycoproteins (GPs), and a normal quantity of large-molecular-weight, multimeric von Willebrand factor (vWF) (Figs. 84-1 and 84-2). Main platelet GPs are GPIb, the platelet receptor for vWF, involved in platelet adhesion, and GPIIb/IIIa, the platelet receptor for fibrinogen, involved in platelet aggregation. Under static conditions, GPIb and vWF have no affinity for each other. However, these molecules develop a specific affinity for each other at high shear stress, resulting in arterial platelet adhesion. Aggregated fibrinogen-platelet mesh acts as a trap for binding and activation of other plasma clotting factors. The exposure of the preceding clotting factors to tissue factors, present on damaged endothelial cells, catalyzes the conversion of prothrombin to thrombin, which converts fibrinogen to fibrin. Subsequent cross-linking of insoluble fibrin results in a stable hemostatic plug. Uremic bleeding caused by acquired platelet dysfunction is a major cause of morbidity and mortality in end-stage renal disease (ESRD) patients. The bleeding problems are characterized by abnormal prolongation of bleeding time and hemorrhagic symptoms, manifesting usually as ecchymoses or petechiae in the skin, epistaxis, gastrointestinal or gingival oozing, or prolonged hemorrhage from needle puncture or postoperative sites. The cutaneous bleeding time (normal values are 1 to 7 minutes) is the best laboratory hallmark of clinical bleeding caused by uremia; however, it is very difficult to standardize and thus not widely used. Other coagulation screen tests, such as activated partial thromboplastin time (APTT), prothrombin time, and thrombin time, are generally normal in patients with uremia.1 Hemorrhagic complications in ESRD patients may also manifest as hemorrhagic pericarditis and/or hemorrhagic pleural effusion as well as intracranial, retroperitoneal, ocular, or uterine bleeding. The pathogenesis of platelet dysfunction in uremia is multifactorial and includes diminished adherence of platelets to vascular endothelium (Box 84-1). In uremic patients, total platelet GPIb content is reduced, whereas the total content of GPIIb and GPIIIa is normal. Platelet membrane GPIb levels and GPIIb/IIIa expression have been described as normal, decreased, or increased in uremic platelets. Plasma vWF level is normal or elevated in uremia. However, the observation that the administration of agents that increase plasma vWF or the factor VIII–vWF complex results in a shortening of prolonged bleeding time and a transient reversal of bleeding tendency in uremia suggests abnormalities in vWF metabolism, structure, and/or function. A variety of circulating plasma proteases, such as plasmin, cleave the α chain of platelet GPIb. The resulting proteolytic degradation product glycocalicin contains binding sites for thrombin and vWF. Elevated plasma glycocalicin levels in uremic patients may contribute to diminished binding of uremic platelets to subendothelium because of the vWF binding site of glycocalicin. Furthermore, glycocalicin contains the thrombin binding site of GPIb which may also contribute to diminished platelet function in uremia. Low GPIb expression on resting platelets obtained from patients with chronic kidney disease (CKD) correlates with the severity of impaired kidney function. However, GPIb expression in uremic platelets increases after stimulation. In contrast, GPIIb/IIIa expression on resting uremic platelets is normal but reduced after stimulation, indicating hyporesponsiveness of the uremic platelets. The ability to bind both vWF and fibrinogen to GPs is reduced in uremia because of a conformational change in the GPIIb/IIIa receptor. Improvement of fibrinogen binding to GPIIb/IIIa by hemodialysis (HD) treatment suggests that uremic toxins such as methylguanidine, guanidinosuccinic acid, phenolic acid, and hydroxyphenylacetic acid also contribute to platelet dysfunction. HD and peritoneal dialysis (PD) correct platelet abnormalities partially because both procedures do not remove all uremic toxins effectively. PD is more effective than HD in improving impaired platelet function and prolonged bleeding time in uremia, possibly because of better clearance of uremic middle molecules. In addition, platelet contact with older cellulose-based HD membranes caused defects in platelet membrane receptors for vWF and fibrinogen, thus preventing normal platelet-vessel wall and platelet-platelet interactions. This can be demonstrated by a more prolonged bleeding time, a more decreased platelet responsiveness to thrombin, and a decreased platelet agglutination in response to ristocetin immediately after the HD procedure. Heparin administered during HD treatment contributes to the prolonged bleeding time immediately after dialysis although not for the platelet alterations described earlier. Platelet contact with the artificial surfaces may result in GPIb internalization, which may also occur during platelet activation. Molecules such as prostacyclin and nitric oxide (NO) inhibit platelet function, modulate vascular tone, affect platelet–vessel wall as well as platelet-platelet interactions, and have an important role in preventing platelet activation under normal conditions. However, uremic platelets generate more NO than platelets obtained from healthy patients, which may contribute to the increased bleeding time in uremia. Uremic plasma induces NO synthesis in endothelial cells. Bleeding tendency in uremic patients is related to the increased platelet nitric oxide synthase (NOS) activity. The NOS substrate l-arginine inhibits platelet aggregation, whereas the NOS inhibitors NG-monomethyl-l-arginine (l-NMMA) and NG-nitro-l-arginine methylester (l-NAME) restore platelet adhesion and aggregation. Inhibition of NOS by l-NMMA restores the increased bleeding time in experimental uremia to normal. Elevated arginine transport in uremic platelets is mediated by the high-affinity amino acid transport system y+L. Thus, increased l-arginine influx into uremic platelets is sustained and may contribute to platelet dysfunction even in the l-arginine–deprived uremic milieu. Renal anemia is another determinant of the prolonged bleeding time in ESRD patients. Within the normal circulation red blood cells increase platelet–vessel wall contact by displacing platelets away from the axial flow and toward the vessel wall. Red blood cells also improve platelet function by releasing adenosine diphosphate (ADP) and inactivating prostacyclin. Vasodilating effects of prostacyclin and NO increase vessel luminal diameter and decrease peripheral dispersion of platelets and their contact with the vessel wall. Recombinant human erythropoietin (rHuEPO) improves platelet function mainly through increasing hematocrit, whereas an increase in blood viscosity may enhance the risk for thrombotic events. Platelet aggregation in uremia is ameliorated by rHuEPO therapy even at a dose that does not influence hemoglobin and hematocrit by the release of young platelets into the blood. Low-dose rHuEPO therapy in patients with uremia also improves impaired platelet aggregation stimulated by ADP and ristocetin. Moderate thrombocytopenia (80 to 150 × 109/l) is a common finding in ESRD patients. The platelet number is reduced in 16% to 55% of uremic patients. However, the platelet count is rarely less than 80 × 109/L, a platelet number generally considered adequate for normal hemostasis. In the past, the interaction of blood with cellulose-based dialyzer membranes (e.g., cuprophane) resulted in complement activation and transient thrombocytopenia during the dialysis procedure. In contrast, relevant platelet reduction does not occur with the use of newer non–complement-activating dialyzer membranes. Heparin may induce thrombocytopenia via the emergence of antibodies. The principle antigen is a complex of platelet factor 4 and heparin (PF-4/H). Frequencies of anti–PF-4/H antibodies range from 0% to 12% in HD patients continuously exposed to heparin. In patients with heparin-induced thrombocytopenia (HIT) type II, platelet counts range from 50 to 80 × 109/l (see later). Increased numbers of circulating reticulated platelets in uremia suggest shortened platelet survival under these conditions. Reduced platelet half-life and low-normal platelet number in uremia suggest increased platelet turnover. Normal controls have a mean of 2.8% ± 0.2% reticulated platelets (a marker of platelet turnover), whereas PD and HD patients have a significantly higher percentage of 6.9% ± 0.7% and 8.2% ± 0.4%, respectively, suggesting enhanced platelet turnover in uremia. Shortened platelet survival in uremia may be the result of increased exposure of negatively charged phosphatidylserine. This signal is recognized by macrophages and promotes phagocytosis of the platelets. The bleeding disorder of uremic patients has been classically defined as an acquired defect of primary hemostasis, characterized by prolongation of bleeding time. Therapeutic strategies include both prevention of bleeding in patients at high risk because of invasive procedures or surgery, and treatment of patients with active bleeding. Several options are available to prevent or treat uremic bleeding. Adequate dialysis (Kt/V > 1.2 in HD and Kt/V > 1.7 in PD patients; see Chapter 94) by removal of uremic toxins improves platelet functional abnormalities. Hemorrhagic problems are fewer with PD than with HD. Deep-tissue biopsies or invasive surgical procedures that require improved hemostasis should ideally be scheduled 12 to 24 hours after dialysis in HD patients. The residual anticoagulant effect of unfractionated heparin (UFH) used during HD lasts as long as 2.5 hours or even longer with the use of low-molecular-weight heparin (LMWH). In case of clinical urgency or high risk of postoperative bleeding, heparin use should be minimized, stopped (using predilutional saline, 100 to 200 ml every 15 or 30 minutes, and a dialysis membrane with low thrombogenicity such as polysulfone), or replaced by regional anticoagulation with citrate during HD. Protamine sulfate administration (1 mg/100 U of heparin infused over 10 minutes) should be considered if there is marked, HD-induced prolongation of the partial thromboplastin time (PTT) and severe bleeding. The severity of anemia in uremic patients correlates with the prolongation of bleeding time. Beneficial effects of red cell transfusion on prolonged bleeding time are independent of changes in platelet function tests or in the level of vWF. Erythropoiesis-stimulating agent (ESA) therapy improves uremic bleeding tendency by several mechanisms (Box 84-2),2 and this has diminished hemorrhagic problems in uremic patients. Increased intraoperative bleeding complications have been reported in ESRD patients with low preoperative hematocrit levels. Unfortunately, in ESRD patients, intraoperative transfusion of packed red blood cells may cause or aggravate hyperkalemia. If surgery is elective, ESA therapy (in combination with intravenous iron) may be administered preoperatively to raise hemoglobin and hematocrit to the upper acceptable values (see Chapter 83). In uremic patients on ESA therapy, a threshold hematocrit of 27% to 32% effectively normalized bleeding time.1
Other Blood and Immune Disorders in Chronic Kidney Disease
Platelet Dysfunction and Coagulation Defects
Bleeding Diathesis in Uremia
Platelet Dysfunction
Platelet Number in Uremia
Therapeutic Strategies
Dialysis
Correction of Anemia
Cryoprecipitate
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Other Blood and Immune Disorders in Chronic Kidney Disease
Chapter 84
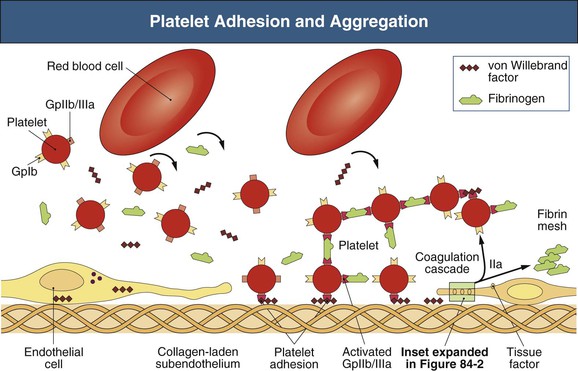
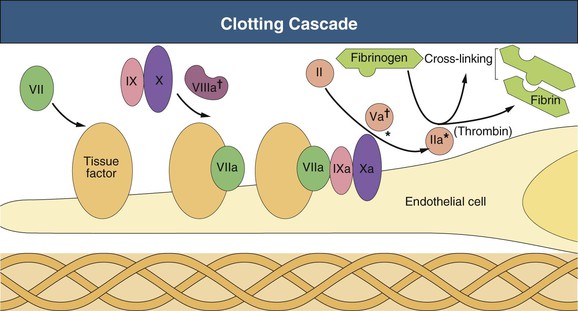
Figure 84-2 Clotting cascade. Expansion of the inset in Figure 84-1 shows the clotting cascade that takes place at the damaged vessel wall. Exposure of subendothelial tissue factor, present on pericytes and fibroblasts, allows eventual activation of prothrombin (factor II) to thrombin. Thrombin converts fibrinogen to fibrin, activates fibrin cross-linking, stimulates further platelet aggregation, and activates anticoagulant protein C. Naturally occurring anticoagulants antithrombin III, protein C, and protein S help maintain control and counterbalance on coagulation. *Site of anticoagulant effect for antithrombin III. †Site of anticoagulant effect for protein C–protein S complex. (Courtesy James A. Sloand, MD, FACP, FASN, Baxter Healthcare Corporation, USA.)






