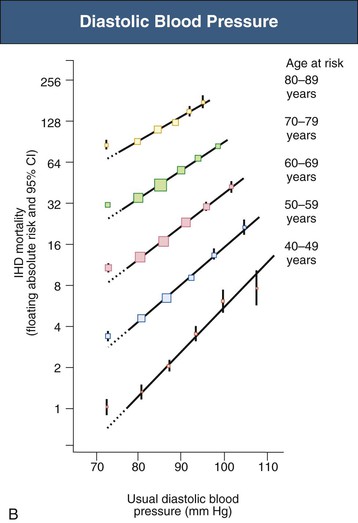
William J. Lawton, Gerald F. DiBona, Ulla C. Kopp, Friedrich C. Luft Systemic arterial blood pressure (BP), or the pressure of the blood within the arteries exerted against the arterial wall, is produced by the contraction of the left ventricle (producing blood flow) and the resistance of the arteries and arterioles. Systolic blood pressure (SBP), or maximum BP, occurs during left ventricular systole. Diastolic blood pressure (DBP), or minimum BP, occurs during ventricular diastole. The difference between SBP and DBP is the pulse pressure.1 The mean arterial pressure (MAP) is clinically defined as the DBP plus one third of the pulse pressure. Blood flow, Q, as defined by Ohm’s law, varies directly with the change in pressure, P, across a blood vessel and varies inversely with the resistance, R, defined as Q = P/R. Rearrangement shows that pressure varies directly with blood flow and resistance, P = QR. Ohm’s law suffices for an overall view of the circulation. However, for a more detailed picture of the resistance to flow in any given vessel, the relationship of Hagen-Poiseuille should be applied, as follows: Here, r is the radius of the pipe, L is its length, and η is the coefficient of viscosity. Thus, as the lumen of a vessel decreases, the pressure increases to the fourth power of the radius for the same blood flow. In other words, a 50% reduction in radius results in a 16-fold increase in pressure to maintain equivalent flow. Normal BP is controlled by cardiac output and the total peripheral resistance and is dependent on the heart, the blood vessels, the extracellular volume, the kidneys, the nervous system, humoral factors, and cellular events at the membrane and within the cell (Fig. 33-1). Cardiac output is determined by the stroke volume in liters per minute (l/min) and the heart rate. In turn, stroke volume is dependent on intravascular volume regulated by the kidneys as well as on myocardial contractility. Myocardial contractility involves sympathetic and parasympathetic control of heart rate, intrinsic activity of the cardiac conduction system, complex membrane transport and cellular events requiring influx of calcium that lead to myocardial fiber shortening and relaxation, and effects of humoral substances (e.g., catecholamines) on stimulating heart rate and myocardial fiber tension. Total peripheral resistance is regulated by baroreflexes and sympathetic nervous system (SNS) activity, response to neurohumoral substances and endothelial factors, myogenic responses, and intercellular events mediated by receptors and mechanisms for signal transduction.2 Baroreflexes are derived from (1) high-pressure baroreceptors in the aortic arch and carotid sinus and (2) low-pressure cardiopulmonary baroreceptors in ventricles and atria. Aortic baroreceptor nerve fibers travel via the vagus nerve (cranial nerve X), and carotid sinus fibers travel via the glossopharyngeal nerve (cranial nerve IX). These receptors respond to stretch (high pressure) or filling pressures (low pressure) and send tonic inhibitory signals to the brainstem. If BP increases and tonic inhibition increases, inhibition of sympathetic efferent outflow occurs and decreases vascular resistance and heart rate. However, if BP decreases, less tonic inhibition occurs, and heart rate and peripheral vascular resistance (PVR) increase, thereby increasing BP. The brainstem cardiovascular centers are localized in the dorsomedial medulla. Neural afferents from cranial nerves IX and X are integrated in the nucleus tractus solitarius (NTS). From here, vasoconstriction and increased heart rate are mediated through the caudal and rostral ventrolateral medulla by the SNS. NTS efferents communicate with the nucleus ambiguus (vagal nucleus) to decrease heart rate through the vagus nerve. Also, the central neural control of renal function modulates renal blood flow, glomerular filtration rate (GFR), excretion of sodium and water, and renin release. These factors in turn regulate intravascular volume, vascular resistance, and BP.3 Inhibitory reflexes also originate in the kidney. Increases in urine flow rate increase renal pelvic pressure, which stretches the renal pelvic wall, leading to activation of mechanosensory nerves in the renal pelvic wall. Activation of these sensory nerves decreases renal sympathetic nerve activity and induces diuresis and natriuresis, an inhibitory renorenal reflex response.4 The responsiveness of the renal sensory nerves is modulated by dietary sodium. A high sodium intake enhances the responsiveness of the afferent renal mechanosensory nerves in rats, thereby achieving sodium balance during increased dietary sodium intake.5 Rats lacking intact afferent renal innervation only achieve sodium balance at the cost of increased MAP. Afferent renal denervation leads to salt-sensitive hypertension.6 Numerous vasoactive substances have effects on blood vessels, the heart, the kidneys, and the central nervous system (CNS) to regulate BP (Table 33-1). The renin-angiotensin-aldosterone system (RAAS) regulates volume and PVR (Fig. 33-2). Angiotensin II (Ang II) constricts vascular smooth muscle; stimulates aldosterone secretion; potentiates SNS activity; stimulates salt and water reabsorption in the proximal tubule; stimulates prostaglandin, nitric oxide (NO), and endothelin release; increases thirst; and stimulates vascular remodeling and inflammation. Increased renal angiotensin activity has also recently been linked with salt-sensitive hypertension, even in the volume-expanded state, and may have a role in blocking pressure natriuresis, in part by increasing renal sympathetic nerve activity and local oxidative stress.7 Aldosterone stimulates sodium channels in distal renal tubular epithelium, leading to sodium retention and potassium excretion. It also exerts inflammatory and fibrotic effects through the mineralocorticoid receptor in vascular cells and in the heart. Plasma concentrations of renin and aldosterone are both inversely related to salt intake and are influenced by various medications. Table 33-1 Vasoactive substances that modulate blood pressure. A second major effector system is the SNS. Sympathetic nerve endings release the vasoconstrictor (norepinephrine) that binds the α-adrenergic receptor (adrenoceptor) on vascular cells, renal cells, and other cells (e.g., adipocytes). Epinephrine increases heart rate, stroke volume, and SBP through α- and β-adrenoceptors. The hormone is released from the adrenal medulla. Increased sympathetic tone has long-term influences on cardiovascular regulation and may cause hypertension.8 In the kidneys, sympathetic nerves mediate renin release. Furthermore, innervation of each individual nephron affects sodium reabsorption. Thus, the SNS regulates both effective circulating fluid volume and PVR. The kallikrein-kinin system counters the RAAS and produces vasodilator kinins, which stimulate prostaglandins and NO (see Fig. 33-2). Prostaglandin E and prostacyclin block the vasoconstriction by Ang II and norepinephrine. Two endothelium-derived factors have opposite effects on the blood vessels: NO is a vasodilator, whereas the endothelins are vasoconstrictors. Natriuretic peptides that include atrial (ANP), brain (BNP), and C-type induce vasodilation and natriuresis and inhibit other vasoconstrictors (renin-angiotensin system [RAS], SNS, endothelin). Renalase is a flavin adenine dinucleotide–dependent amine oxidase that is secreted by the kidney, circulates in the blood, and modulates cardiac function and systemic BP by metabolizing catecholamines.9 Heme oxygenase (HO) is another important intrarenal and systemic BP modulator and inhibits oxidants, resulting in decreased BP.10 Endogenous digitalis-like factors, which inhibit cell surface Na,K-ATPase and include an ouabain-like factor and marinobufagenin, also appear to regulate BP, cardiovascular (CV), and renal function.11 Urotensin II is a locally expressed vasoconstrictive cyclic vasoactive peptide that stimulates proliferation of vascular smooth muscle cells and fibroblasts, inhibits insulin release, and modulates GFR. Despite urotensin II having these actions, high plasma urotensin II levels predict reduced CV complications in patients with chronic kidney disease (CKD, stages 2 to 5).12,13 Other mediator systems recently discovered include leptin, which in obese persons may increase BP by activating the CNS through a melanocortin pathway.14 Uric acid may also be important by activating the RAS, inducing intrarenal oxidative stress, blocking endothelial NO, and directly affecting the vasculature.15 Recent studies suggest a role for T cells in primary hypertension, possibly in response to heat shock protein 70, which may cause intrarenal vasoconstriction through release of Ang II and oxidants.16 Chemoreceptors in the brain medulla and in the carotid and aortic bodies respond to changes in carbon dioxide and oxygen tension, resulting in renal vasoconstriction and dilation of the CNS and coronary vasculature.17 Pain can also activate the SNS, although deep pain from crush injuries, testicular trauma, joint disruption, or abdominal distention leads to decreased sympathetic and increased parasympathetic outflow and decreased BP.17 Postreceptor signaling events also regulate PVR. For example, the small guanosine triphosphatase Rho and its effector, Rho-associated kinase (Rho-kinase), stimulate vascular constriction and may have a role in cerebral artery spasm.18 Guyton and Hall1 provide a model of the BP regulation in which CNS mechanisms (e.g., baroreflexes) provide short-term regulation of the circulation (seconds to minutes), with the RAAS and fluid shifts altering BP over minutes to hours and the kidneys responsible for long-term adjustments in BP, predominantly by regulating extracellular fluid volume1 (Fig. 33-3). Indeed, the critical role for the kidney in the long-term control of BP received strong support from kidney cross-transplantation experiments19 and bilateral nephrectomy in patients with end-stage renal disease (ESRD) and severe hypertension. However, activation of CNS and renal SNS pathways are now recognized as important in long-term BP control, in part by the later effects on the kidneys.20 Renal sympathetic denervation by radiofrequency ablation through the renal arteries leads to substantial reduction in blood pressure in patients with resistant hypertension (see Chapter 38).21,22 Furthermore, both intrarenal and extrarenal mechanisms are involved in the long-term regulation of blood pressure.23 Blood pressure is normally distributed in the general population. Thus any definition of hypertension is arbitrary. Hypertension is usually asymptomatic, and most symptoms, if present, are more from the sequelae of hypertension or its treatment. Hypertension may be defined by its associated morbidity and mortality, as increases over arbitrary cutoff points, or by thresholds defining therapeutic benefit. The first approach defines hypertension by relating BP levels to the risk of morbidity and mortality. The association of SBP and DBP with CV and renal complications is continuous over the entire BP range.24 Death from both coronary heart disease and cerebrovascular accident (stroke) increases progressively and linearly from BP as low as 115 mm Hg SBP and 75 mm Hg DBP upward in all age groups ranging from 40 to 89 years (Figs. 33-4 and 33-5). For every increase of 20 mm Hg SBP or 10 mm Hg DBP, the mortality from both coronary heart disease and stroke doubles. On the basis of these data, the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7 Report) introduced the term prehypertension for those with BP ranging from 120 to 139 mm Hg SBP or 80 to 89 mm Hg DBP.25 After the 2003 JNC 7, the Writing Group of the American Society of Hypertension (WG-ASH) proposed a new definition of hypertension not based on BP values alone but considering hypertension as a complex cardiovascular disorder that includes target organ damage, early disease markers, and CV risk factors26 (Table 33-2). This risk-based approach seeks to identify individuals with an increased likelihood of future CV events at any BP level. The WG-ASH stage 1 includes the prehypertension category in JNC 7. It should be noted that JNC 8 has recently been published but focuses on BP management and not on classification.26a Table 33-2 Working Group–American Society of Hypertension (WG-ASH) definition and classification of hypertension. * Cardiovascular (CV) disease determined by constellation of risk factors, early disease markers, and target organ disease. A second approach defines hypertension by the frequency distribution within a population. This statistical approach arbitrarily designates values above a certain percentile as “hypertensive” and is used in defining hypertension in children. The values defining hypertension vary according to age, gender, body size, and race.27 This frequency distribution method is not helpful for determining a value for initiation of antihypertensive treatment but is useful in epidemiologic studies, for example, defining the prevalence of hypertension in various age groups or the changing prevalence of hypertension in a given population over time. The prevalence of hypertension in adults in the United States, defined as BP of 140/90 mm Hg or higher, has increased progressively from 11% of the population in 1939 to 29.3% in 2004.28 The reported prevalence of hypertension in six European countries is 44%, and 66.3% in those older than 60.29 The third concept for defining hypertension is derived from randomized controlled trials (RCTs) that have demonstrated reductions in mortality and morbidity. As a result of clinical RCTs, consensus has been reached on intervention levels for moderate and severe hypertension but not for lower levels of hypertension. The Hypertension Optimal Treatment (HOT) study showed benefits of lowering BP to 138/83 mm Hg in hypertensive patients in the absence of diabetes mellitus, CKD, coronary heart disease risk, or target organ damage.30 For patients with these complications, a treatment goal below 130/80 mm Hg is recommended (JNC 7).25 The European Society of Hypertension (ESH) divides “normotension” into three categories (optimum, normal, high-normal) and describes hypertension as mild, moderate, or severe31 (Table 33-3). The ESH also provides values for automated 24-hour BP measurements, divided into daytime and nighttime. Table 33-3 European Society of Hypertension consensus classification. Blood pressure readings are in millimeters of mercury (mm Hg). * Consensus classification proposed by the European Society of Hypertension/European Society of Cardiology guidelines (see reference 32). † Classification proposed at the Eighth International Consensus Conference on Ambulatory Blood Pressure Measurement (Sendai, Japan, October 2001). ‡ Mean of 95th percentiles in patients who on conventional BP measurement were normotensive in large-scale studies. In the United States, JNC 7 defined hypertension for individuals 18 years and older.25 The committee settled on normal, prehypertension, and stage 1 and stage 2 hypertension (Table 33-4). For children, JNC 7 considers that BP at the 95th percentile or higher at each age is elevated. Table 33-4
Normal Blood Pressure Control and the Evaluation of Hypertension
Normal Blood Pressure Control
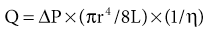
Vasoactive Substances that Modulate Blood Pressure
Group
Compound
Cellular Effects
Catecholamines
Norepinephrine, epinephrine, dopamine
Adrenergic receptors (α1, α2, β1, β2) causing protein phosphorylation and increased intracellular calcium through G proteins linked to ion channels or second messengers (cyclic nucleotides, phosphoinositide hydrolysis)
Renin-angiotensin system (RAS)
Angiotensin II (Ang II)
Angiotensin receptors (AT1, AT2, AT4) causing increased intracellular calcium and protein phosphorylation through second messenger, phosphoinositide hydrolysis, and activated protein kinases
Aldosterone stimulation
Mineralocorticoids
Aldosterone
Genomic: Binds to cytoplasmic mineralocorticoid receptor (MR), translocates to nucleus, modulates gene expression, and signals transduction and effectors (SgK, CHIF, Ki-Ras), which increases transport proteins (increasing ENaC number and open probability)
Nongenomic: Effects through separate membrane or cytosolic proteins
Kallikrein-kinin system
Bradykinin
Bradykinin receptors (B1, B2), B2-G protein coupling cause activation of phospholipase C, increased inositol phosphates, and intracellular calcium
Arachidonic acid products
Prostaglandins: prostaglandin E, prostacyclin, thromboxanes
Lipoxygenase enzyme products: leukotrienes, hydroxyeicosatetraenoates
Nine prostaglandin receptors coupled to G proteins: (e.g., PGI2 [Receptor IP], PGE2 [Receptors EP1, EP2]); PGF2a (Receptor FP)
Endothelium-derived factors
Endothelium-derived relaxing factor (nitric oxide)
Endothelins (ET-1, ET-2, ET-3)
Increased levels of cyclic guanosine monophosphate cause activation of protein kinases
G proteins activate phospholipase C and l-type calcium channels
Class 2 G protein–coupled receptor
Natriuretic peptides
Atrial, brain, and C-type
Activation of three receptor types; further effects mediated by cGMP
Posterior pituitary hormones
Arginine vasopressin
Vasopressin receptors (AVPR 1A; AVPR 1B) mediated by second messenger system, phosphatidyl inositol/calcium; AVPR2 effects via adenylate cyclase (cAMP)
Cyclic vasoactive peptides
Urotensin II (UT II)
UT II binds to G protein receptor GPR 14
Other substances
Acetylcholine, adenosine, insulin, neuropeptide Y, serotonin, sex hormones (estrogens, progesterone, androgens), glucocorticoids, other mineralocorticoids, substance P, vasopressin, renalase, heme oxygenase 1
Definition of Hypertension
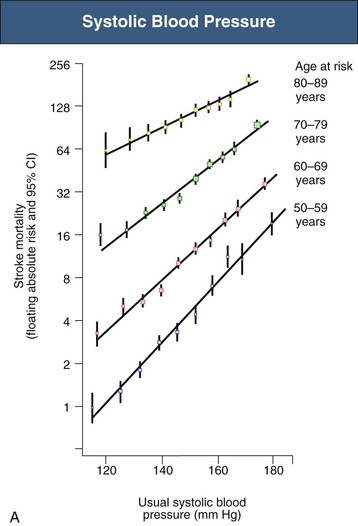
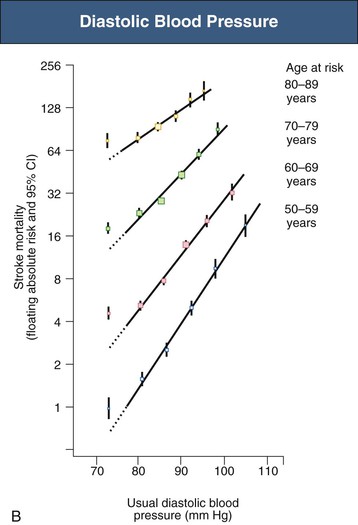
Blood Pressure in Relation to Morbidity and Mortality
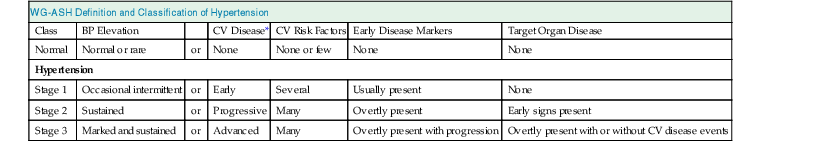
WG-ASH Definition and Classification of Hypertension
Class
BP Elevation
CV Disease*
CV Risk Factors
Early Disease Markers
Target Organ Disease
Normal
Normal or rare
or
None
None or few
None
None
Hypertension
Stage 1
Occasional intermittent
or
Early
Several
Usually present
None
Stage 2
Sustained
or
Progressive
Many
Overtly present
Early signs present
Stage 3
Marked and sustained
or
Advanced
Many
Overtly present with progression
Overtly present with or without CV disease events
Elevation of Blood Pressure by Arbitrary Cutoff Points
Threshold of Therapeutic Benefit
Operational Definitions
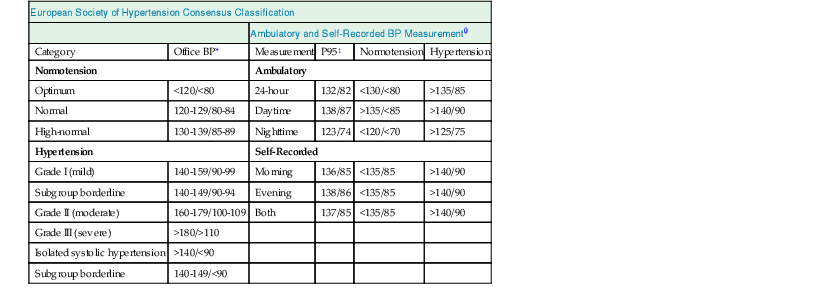
European Society of Hypertension Consensus Classification
Ambulatory and Self-Recorded BP Measurement†
Category
Office BP*
Measurement
P95‡
Normotension
Hypertension
Normotension
Ambulatory
Optimum
<120/<80
24-hour
132/82
<130/<80
>135/85
Normal
120-129/80-84
Daytime
138/87
>135/<85
>140/90
High-normal
130-139/85-89
Nighttime
123/74
<120/<70
>125/75
Hypertension
Self-Recorded
Grade I (mild)
140-159/90-99
Morning
136/85
<135/85
>140/90
Subgroup borderline
140-149/90-94
Evening
138/86
<135/85
>140/90
Grade II (moderate)
160-179/100-109
Both
137/85
<135/85
>140/90
Grade III (severe)
>180/>110
Isolated systolic hypertension
>140/<90
Subgroup borderline
140-149/<90
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Normal Blood Pressure Control and the Evaluation of Hypertension
Chapter 33
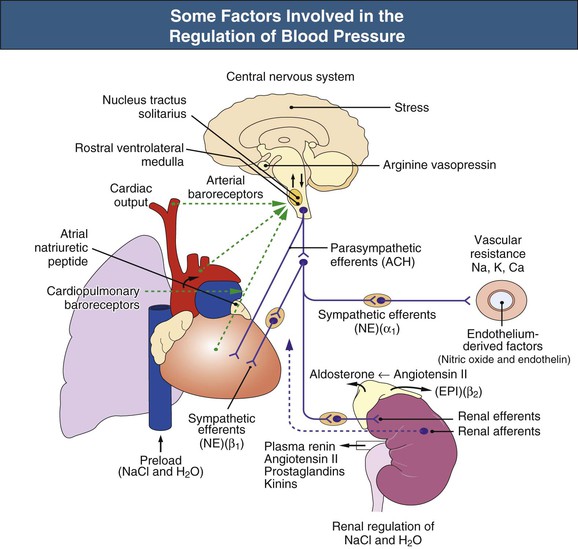
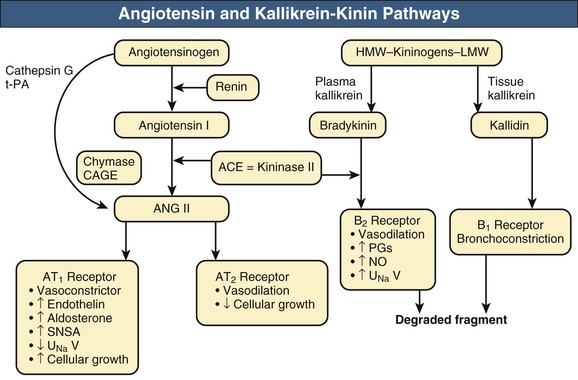
Figure 33-3 Temporal sequence for adjustment of blood pressure control. Degree of activity, expressed as feedback gain, of several arterial blood pressure control systems at various times after a sudden change in arterial BP. Note the infinite gain of the renal-volume mechanism for BP control. CNS, Central nervous system. (From reference 1.)
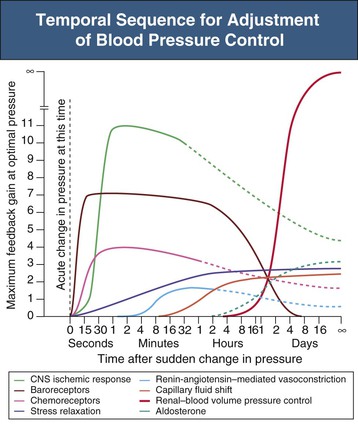
Figure 33-4 Ischemic heart disease (IHD) mortality rate in each decade of age versus usual blood pressure at start of that decade. A, Systolic blood pressure. B, Diastolic blood pressure. Floating absolute risk is a relative risk score that corrects for a baseline group, in this case the absolute death rate within a particular community. The size of the squares correlates inversely with the variance of the data collected for that data point. CI, Confidence interval. (From reference 24.)
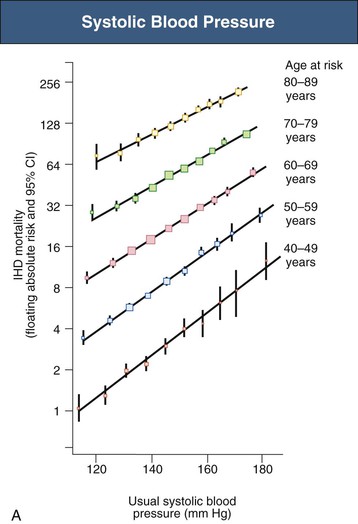
Figure 33-5 Stroke mortality rate in each decade of age versus usual blood pressure at start of that decade. A, Systolic blood pressure. B, Diastolic blood pressure. Floating absolute risk is a relative risk score that corrects for a baseline group, in this case the absolute death rate within a particular community. The size of the squares correlates inversely with the variance of the data collected for that data point. CI, Confidence interval. (From reference 24.)






