Nephrotoxicity Secondary to Environmental Agents, Heavy Metals, Drug Abuse, and Lithium
Veronica Torres da Costa e Silva
Etienne Macedo
Luis Yu
Environmental kidney diseases are a consequence of occupational exposure, a particular form of environmental disease. Recognized chronic occupational renal diseases include those caused by exposure to heavy metals, organic solvents (aliphatic, aromatic, and halogenated hydrocarbons), and silica. Minamata disease from mercury and Itai-Itai disease from cadmium are two environmental diseases caused by industrial elements. Another disease is Balkan nephritis presumed to be of environmental origin and the nephropathy caused by the ingestion of germanium compounds.
Several heavy metals are generally recognized as nephrotoxic following environmental or occupational exposure, including: lead, cadmium, mercury, uranium, chromium, copper, and arsenic. However, chronic renal failure has been described only for lead, mercury, cadmium, uranium, and arsenic. Therapeutic forms of platinum, gold, lithium, and bismuth may also induce kidney damage and these aspects are explored in other chapters of this book. Other heavy metals with potentially nephrotoxic effects are barium, cobalt, manganese, nickel, silver, thallium, thorium, tin, and vanadium but there is no definitive evidence they can actually lead to renal disease.
URINARY BIOMARKERS
Urinary proteins and biochemical markers were associated with toxic renal injury. Urinary biomarkers may reflect specific sites of renal injury: (1) low molecular weight proteins and intracellular enzymes—proximal tubule damage; (2) Tamm-Horsfall glycoprotein and kallikrein—distal tubule injury; (3) high molecular weight proteins—increased glomerular permeability (if >200 mg per g creatinine); and (4) biochemical markers—eicosanoids suggesting vascular injury.
A group of urinary markers (human intestinal alkaline phosphatase [HIAP], total nonspecific alkaline phosphatase [TNAP], N-acetyl-β-D-glucosaminidase [NAG], retinol binding protein [RBP], Tamm-Horsfall glycoprotein [THG], β2– microglobulin, microalbumin, thromboxane B2 [TBX2], and three prostaglandins [prostaglandin E2, PGE2; prostaglandin F2α, PGF2α; 6-keto-prostaglandin F1α, 6-keto-PGF1α]) are detailed to demonstrate the differentiation of the toxic nephropathies (Table 34.1).1,2,3,4,5
The excretion patterns represent occupational exposure levels indicated by the specified mean blood or urine concentrations. Mercury and cadmium exposure3,4,5 is associated with the isoenzyme HIAP, a sensitive and specific indicator of injury to the S3 segment of the proximal tubule. Total nonspecific alkaline phosphatase is increased after perchloroethylene exposure6,7 and NAG and RBP are elevated after cadmium exposure.3,8 The urinary eicosanoids PGE2, PGF2α, and 6-keto-PGF1α seem to be associated to the development of hypertension and injury to the glomeruli or renal medulla. Also, low levels of urinary albumin can also express proximal tubular dysfunction and the failure to reabsorb or metabolize albumin that passes through the glomerular filter.
The ability of these urinary markers to discriminate among the diverse nephrotoxins enhances with increasing exposure levels. Urinary markers should be collected in fasting fresh voided specimens (spot urines) at 8 AM, and expressed relative to the creatinine concentration. The specificity of tubular injury decreases in the presence of renal damage.
LEAD NEPHROPATHY
The first description of lead nephrotoxicity was made by Lancereaux in 1862, who reported a patient with saturnine (lead-induced) gout with kidneys showing interstitial nephritis at postmortem examination. Nevertheless, the demonstration of lead nephrotoxicity has presented some issues. There are difficulties proving the connection between late sequelae of chronic absorption to relatively low levels of lead, and distinguishing between glomerular and extraglomerular renal disease.9 Another difficult aspect is how to separate the transient Fanconi syndrome of acute childhood lead poisoning from the chronic interstitial nephritis characteristic of lead nephropathy in adults. Finally, one confounding aspect is the differentiation of late complications of excessive lead
absorption and gout and hypertension renal lesions, which are other important causes of renal disease.
absorption and gout and hypertension renal lesions, which are other important causes of renal disease.
TABLE 34.1 Urinary Markers in Toxic Nephropathies—European Cooperative Study | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Diagnosis
The mainstay of laboratory diagnosis is the blood lead concentration, which is usually over 60 µg per dL, although recent evidence of lead-induced organ damage occurred with blood levels over 10 µg per dL.10 Blood lead concentrations usually decrease significantly within 4 weeks of removal from exposure making the blood lead concentration relatively insensitive to cumulative body stores acquired over years. Around 95% of the body stores of lead are accumulated in the bone with a mean residence time of approximating 20 years11 and cumulative past lead absorption is best assessed by the calcium disodium edetate (CaNa2EDTA) lead-mobilization test.12
The ethylenediamine tetraacetic acid (EDTA) test is performed in adults by parenteral administration of 1 to 3 g (intravenous or intramuscular) of CaNa2EDTA over 4 to 12 hours with subsequent collection of 24-hour urine samples over 1 to 4 days. The intramuscular administration of 2 g of CaNa2EDTA (1 g of EDTA mixed with local anesthetic in each of two injections, 12 hours apart) seems to be a better option of performing the chelation test because it has been well standardized in both normal subjects and patients with renal failure.13,14,15,16,17,18 In the presence of renal damage (serum creatinine > 1.5 mg per dL), urinary excretion of lead chelate should be extended to at least 3 consecutive days and the adequacy of collection checked by simultaneous measurement of urinary creatinine excretion (1.3 g of creatinine per day is an acceptable lower limit in normal adult males). Adults without undue prior lead absorption excrete up to 650 µg of lead-chelate in the urine.
Acute Lead Nephropathy
In acute lead nephropathy, a proximal tubule reabsorptive defect characterized by aminoaciduria, phosphaturia, and glycosuria (Fanconi syndrome) is observed,23 usually in the presence of blood lead levels in excess of 150 µg per dL. An increase in urinary NAG is observed, which correlates positively with the blood lead concentration.24 Tubular dysfunction is often reversed after chelation therapy is initiated to treat the more dangerous encephalopathy. Acute lead nephropathy is associated with acid-fast intranuclear inclusions in proximal tubule epithelial cells25 which are a lead-protein complex, also observed in the urinary sediment.25 These inclusions can occur in other organs such as in liver, neural tissue, and osteoclasts. Morphologic and functional defects in mitochondria are also observed in acute poisoning.
Chronic Lead Nephropathy
Chronic lead nephropathy is the slowly progressive interstitial nephritis observed in adults after prolonged lead exposure. The disease is more frequently recognized in lead workers after long periods (decades) of exposure but other groups have been described such as young adults who sustained acute childhood lead poisoning,26 illicit whiskey (“moonshine”) consumers, U.S. armed service veterans suffering from renal failure attributed to gout or essential hypertension,13,14 and sporadic case reports such as geophagia,27 Asian folk remedies, and cosmetics.
Chronic lead nephropathy causes chronic renal damage less responsive to chelating agents. There is evidence of associated functional impairment such as inhibition of both the renin-angiotensin system and Na+-K+-ATPase,17,28 and these effects may be changed by chelation therapy. This explains why some exposed individuals can restore previous reductions in glomerular filtration rate (GFR) after long-term low-dose chelation therapy (1 g of CaNa2EDTA with local anesthetic three times weekly until the chelation test returns to normal). Proteinuria and glycosuria are initially absent and an increase in TXB2, and a decrease in PGE2 and 6-keto-PGF1α, in the urine is observed.2,24
Renal biopsies in chronic lead nephropathy show nonspecific tubular atrophy and interstitial fibrosis with minimal inflammatory response as well as mitochondrial swelling, loss of cristae, and increased lysosomal dense bodies within proximal tubule cells (Fig. 34.1).15,19 Arteriolar changes indistinguishable from nephrosclerosis are found, often in the absence of clinical hypertension. Intranuclear inclusion bodies are often absent when the renal disease is long-standing or following the administration of chelating agents. Clumped chromatin and nuclear invaginations of cytoplasmic contents may be found even in the absence of intranuclear inclusions. Morphologic alterations are minimal in glomeruli until the reduction in GFR is advanced. This helps explain the association between lead toxicity and hypertension.
The association between lead and gout nephropathy has long been described.29,30 Hyperuricemia and gout are common among individuals with excessive exposure to lead, apparently the result of decreased excretion and increased production of uric acid, and half of uremic patients with lead nephropathy have clinical gout.26 There is substantial evidence that renal failure in gout is sometimes secondary to overt or unsuspected lead poisoning. In Queensland, Australia, as many as 80% of gout patients with renal failure have elevated EDTA lead-mobilization tests.26 In New Jersey chelatable lead was found to be significantly greater among gout patients with renal failure than among gout patients with normal renal function.13 Therefore, unrecognized lead poisoning can be an explanation for renal failure in some gout patients with no evidence of urinary calculi or intratubular uric acid deposition disease.
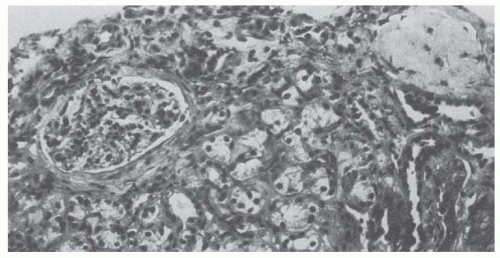 FIGURE 34.1 Renal biopsy obtained from a 28-year-old man who had prepared lead solder for 5 years. His 125I-iothalamate clearance was 52 mL/min/1.73 m2; hemoglobin, 9.6 g per dL; uric acid, 13.2 mg per dL; and blood lead, 48 µg per dL when he was initially seen. Lead-chelate excretion following 2 g of CaNa2EDTA intramuscularly was 5.2 mg for 24 hours. Light microscopy shows periglomerular fibrosis, a sclerotic glomerulus, and tubular atrophy. (Trichrome stain; magnification ×304.) (From Wedeen RP, Maesaka JK, Weiner B, et al. Occupational lead nephropathy. Am J Med. 1975;59:630, with permission.) |
The initial renal injury from lead seems to be in the microvascular endothelium.31 Although the mechanism of injury is not completely clear, it is well known that metabolism of lead is similar to that of calcium and other cations. Also, lead interacts with vasoactive substances that may modulate blood pressure and induce endothelial injury.32,33 These aspects help to explain the association between lead toxicity and hypertension as suggested in several reports.34,35 Some patients presenting as “essential hypertension with nephrosclerosis” may have evidence of lead nephropathy by the EDTA lead-mobilization test.14 Mortality from hypertensive cardiovascular disease is more frequent among lead workers than the general population.36 In these patients, lead seems to contribute to hypertension particularly in the presence of renal dysfunction.17
Treatment
Lead nephropathy is one of the few renal diseases that is preventable and potentially reversible by judicious use of chelation therapy.16,37,38 However, there is no evidence that such therapy reverses established interstitial nephritis especially if serum creatinine concentration exceeds 3 mg per dL.39 Reports of partial remissions may be the reversal of acute poisoning superimposed on chronic lead nephropathy.
Before chelation therapy is undertaken, it may be necessary to perform the EDTA lead-mobilization test and other possible causes of renal disease should be excluded. Long-term, low-dose EDTA therapy should be undertaken until an endpoint is achieved, such as reversion of the EDTA test to normal and restoration of renal function. The cumulative nephrotoxicity of prolonged EDTA therapy in patients with advanced renal failure is unknown. Reports of deterioration of renal function after CaNa2EDTA therapy have been described and treated patients warrant careful follow-up.40,41
CADMIUM NEPHROPATHY
Several compounds containing cadmium are widely used in the manufacturing of pigments, plastics, glass, metal alloys, and electrical equipment. Acute absorption of small quantities as 10 mg of dust or fumes may cause severe gastrointestinal symptoms and fatal pulmonary edema, after a delay of 8 to 24 hours.42 Chronic low dose exposure leads to slowly progressive emphysema, anosmia, and proximal tubular reabsorption defects characterized by low molecular weight proteinuria, enzymuria, aminoaciduria, and renal glycosuria.43,44,45,46 Hypercalciuria (with normocalcemia), phosphaturia, and distal renal tubular acidosis result in clinically important osteomalacia, pseudofractures, and urinary tract stones.47,48 Proximal tubular dysfunction can progress to chronic renal failure over years.49,50
Metabolism
Nonoccupationally exposed individuals can accumulate cadmium through food and cigarettes. The biologic half-life of cadmium in humans exceeds 15 years, and one third of the total body stores (10 to 20 mg) are accumulated in the kidneys.
Absorbed cadmium is initially sequestered in liver and kidney, where it is bound to metallothionein, a cysteine-rich apoprotein.42,43 The cadmium-thionein complex is filtered at the glomerulus, taken up in the proximal tubule by endocytosis, and transferred to lysosomes, where it is rapidly degraded. Most of the cadmium accumulated in the proximal tubules is bound to protein and after a “critical concentration” of 200 µg per g of renal cortex is reached, renal effects become evident. Normal urinary cadmium excretion is usually under 2 µg per day and values over 10 µg per day are associated with cadmium accumulation. Urinary cadmium excretion in excess of 30 µg per day correlates with significant abnormalities of proximal tubular function.51 Although blood cadmium concentration is less reliable as an indicator of health effects and cumulative absorption, blood levels greater than 1 µg per dL are considered evidence of excessive exposure.
β-2 microglobulin has been the most extensively examined urinary protein in cadmium nephropathy. Its excretion is an early renal effect of cadmium42 but, considering its instability in acid urine, measurement of urinary RBP or NAG is probably more reliable.3,8,24,51 Low level of albumin and transferrin are observed in the urine of cadmium workers with low molecular weight proteinuria and enzymuria,3,52 but it is not clear whether this means glomerular injury or impaired tubular reabsorption. Proteinuria in cadmium workers rarely exceeds a few hundred milligrams per day and does not approach nephrotic levels.
Calcium Wasting
The leading feature of cadmium tubular dysfunction is increased calcium excretion.53 Although osteomalacia is uncommon in cadmium workers, it can be observed, associated with diminished renal tubular reabsorption of calcium and phosphate, elevated circulating parathormone levels, and reduced hydroxylation of vitamin D metabolites.54,55 Urinary calculi have been reported in up to 40% of those subjected to industrial exposure and ureteral colic is more likely to be the cadmium worker’s chief complaint.43,47,56
Itai-Itai Disease
Itai-Itai disease is a painful bone condition associated with pseudofractures caused by cadmium-induced renal calcium wasting, first described in Japan. The origin is attributed to local contamination of food staples by river water polluted with industrial effluents, particularly cadmium. The syndrome afflicts postmenopausal, multiparous women presenting with reduced GFR, anemia, lymphopenia, and hypotension as well as osteomalacia. They also exhibit a waddling gait, short stature, anemia, glucosuria, and elevated serum alkaline phosphatase levels. β2-microglobulin urinary excretion exceeds the normal maximum (1 mg per g of creatinine) by almost 100-fold, predicting the later development of renal failure. The renal damage progresses even after exposure has ceased.
Chronic Interstitial Nephritis
Although the role of cadmium in the induction of chronic interstitial nephritis has been controversial, analysis of postmortem tissue or renal biopsy specimens of exposed individuals was able to find tubulointerstitial nephritis.57 These findings associated with recent epidemiologic studies in the United States56 and Belgium,58,59 and the long-term follow-up of Itai-Itai disease in Japan,54 have consolidated cadmium as a cause of chronic interstitial nephritis. These studies presented some important evidence: there was an association between cumulative cadmium exposure and the later increase in serum creatinine after a latent period of several decades; and among exposed workers, there was an increase in serum creatinine concentrations over 5 years accompanied by an important increase in mean urinary β2-microglobulin and loss of glomerular filtration (around 30 mL per minute, 30 times the predicted loss of kidney function for the group).
Diagnosis
Cadmium nephropathy is usually diagnosed based in a history of exposure associated with laboratory tests indicative of proximal tubule injury (e.g., increased excretion of urinary biomarkers, hypercalciuria, or renal glycosuria). Cadmium concentration over 10 µg per g of creatinine confirms the diagnosis. Assessment of renal and hepatic accumulation of cadmium by neutron-activation analysis has been explored and organ cadmium content correlates well with tissue and urinary cadmium levels and β2-microglobulin excretion. When the hepatic cadmium level exceeds about 60 ppm, and renal cortical content exceeds 200 ppm (20 mg per kidney), tubular proteinuria is likely to occur. The diagnostic value of neutron-activation analysis of kidney is decreased in uremic patients because renal cadmium concentration tends to fall with the development of renal failure.59
Treatment
The chelating agent CaNa2EDTA has little effect after cadmium has been complexed with metallothionein60 and it is not usually recommended. Progression of renal disease may occur despite removal from exposure.54 Osteomalacia may be controlled by calcium and vitamin D replacement51 and urinary tract stones are not a contraindication to such therapy.
MERCURY
The toxicity of mercury depends on both its chemical form and the route of absorption. Elemental mercury produces neurologic disease and even death but it does not cause nephrologic damage. However, the mercuric salt corrosive sublimate (HgCl2) is the most nephrotoxic form which is accumulated in the cells of proximal tubules inducing acute tubular necrosis (ATN).61 This mercurial compound binds avidly to sulfhydryl groups in circulating proteins and amino acids as well as intracellular glutathione, cysteine, and metallothionein. Mercury accumulates in the pars recta of proximal tubules which is accomplished by transport primarily from the luminal side of mercury bound to amino acids or proteins. Mercury-ligand complexes reach lysosomes by endocytosis62 with subsequent release into the cytosol by intralysosomal enzymatic degradation.
Diagnosis
The diagnosis of mercury-induced renal disease is usually dependent on known exposure in the presence of renal dysfunction. Although blood mercury levels over 3 µg per dL or urine levels above 50 µg per g of creatinine are considered abnormal, the correlation of blood and urine concentrations with renal disease is poor.63 Mercury exposure is associated with increased HIAP urinary excretion but little increase in TNAP, NAG, RBP, THG, β2 microglobulin, or microalbuminuria (Table 34.1).1




Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
