Nephrotoxicity of Nonsteroidal Anti-inflammatory Agents, Analgesics, and Inhibitors of the Renin-Angiotensin System
Nephrotoxicity of Nonsteroidal Anti-inflammatory Agents, Analgesics, and Inhibitors of the Renin-Angiotensin System
Biff F. Palmer
NEPHROTOXICITY OF NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
Nonsteroidal anti-inflammatory drugs (NSAIDs) are some of the most widely used therapeutic agents in clinical practice today. Although the gastrointestinal toxicity of these medications is well known, it has become increasingly apparent that the kidney is also an important target for untoward clinical events. The renal toxicity associated with the use of NSAIDs can be divided into one of several distinct clinical syndromes. These include a form of vasomotor acute renal failure, nephrotic syndrome associated with interstitial nephritis, chronic renal injury, and abnormalities in sodium, water, and potassium homeostasis. The common link in these syndromes is a disruption in prostaglandin metabolism, the class of compounds whose synthesis is inhibited by these agents.
PROSTAGLANDIN BIOSYNTHESIS AND COMPARTMENTALIZATION
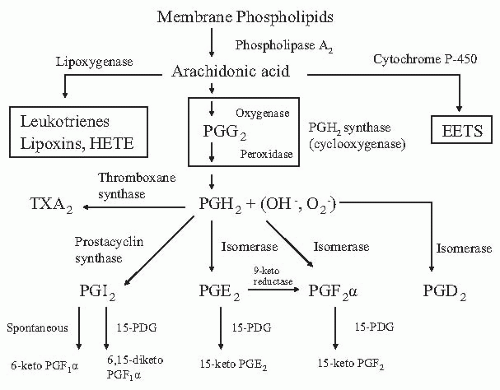
Prostaglandins are members of a class of compounds termed eicosanoids. Eicosanoids are biologically active fatty acids that are all derived from the oxygenation of arachidonic acid. The particular enzyme involved in the oxygenation process dictates which class of eicosanoid will be synthesized. Oxygenation of arachidonic acid by the enzyme cyclooxygenase is responsible for prostaglandin and thromboxane synthesis (Fig. 32.1). The enzyme lipoxygenase converts arachidonic acid to leukotrienes, lipoxins, and eventually, to hydro fatty acid derivatives such as hydroxy eicosatetraenoic acid (HETE). Finally, oxygenation by the cytochrome P-450 system generates epoxyeicosatrienoic acids (EETs).
The availability of free arachidonic acid is the ratelimiting step in eicosanoid biosynthesis. Normally, arachidonic acid is found esterified to membrane phospholipids, where it undergoes deacylation primarily under the influence of phospholipase A2. Phospholipase A2-mediated arachidonic acid release is a calcium-calmodulin-dependent step that is stimulated by vasopressin, bradykinin, angiotensin, and norepinephrine.1 Once released, free arachidonic acid is either re-esterified back into membrane lipids or is converted into one of the biologically active eicosanoids.
The first step in the synthesis of prostaglandins and thromboxanes is a cyclooxygenase reaction in which arachidonic acid is converted into the cyclic endoperoxide prostaglandin G2 (PGG2). PGG2 then undergoes a peroxidase reaction to form a second endoperoxide called PGH2, which is accompanied by the formation of a superoxide radical. Both of these reactions are catalyzed by the enzyme cyclooxygenase (COX), also known as prostaglandin endoperoxide H synthase.2,3 The cyclooxygenase and peroxidase reactions occur on distinct but neighboring sites on the COX enzyme. Once formed, PGH2 has a short half-life and is rapidly acted on by a series of enzymes that produce biologically active prostaglandins or thromboxane. Prostacyclin synthase acts to form prostacyclin (PGI2), thromboxane synthase forms thromboxane A2, and isomerases are responsible for the formation of PGE2, PGD2, and PGF2α.
Prostaglandins are synthesized on demand and exert physiologic effects in discrete microenvironments along the nephron in close proximity to their points of synthesis (Table 32.1). Due to the virtual absence of distant effects, these compounds are best regarded as autacoids rather than hormones. Variations in the synthetic and degradative machinery along the length of the nephron account for the differing types and amounts of prostaglandins found in any given segment.4 PGI2 is the most abundant prostaglandin produced in the cortex and is primarily synthesized in cortical arterioles and glomeruli.5 This location corresponds to the known effects of PGI2 in regulating renal vascular tone, the glomerular filtration rate (GFR), and renin release. PGE2 and thromboxane A2 are also produced in the glomerulus and therefore may exert effects at this site.
The most abundant prostaglandin found in the tubules is PGE2.5 The cortical and especially the medullary portion of the collecting duct are the dominant sites of PGE2 synthesis. Lesser amounts are found in the thin descending and thick ascending limb with the least amount of synthesis found in the proximal tubule. Medullary interstitial cells are also a rich source of PGE2 production. This distribution provides the anatomic basis for PGE2 to modulate sodium and chloride transport in the Henle loop, regulate arginine vasopressin-mediated water transport, and control vasa recta blood flow. PGF2α is synthesized primarily by medullary interstitial cells and less by the papillary collecting tubule and glomeruli. Prostaglandin-degradative enzymes are found in both the cortex and medulla but are most abundant in the cortex. Except for PGI2, which undergoes spontaneous hydrolysis to 6-keto-PGF2α, prostaglandins are rapidly metabolized into inactive products by a 15-prostaglandin dehydrogenase. An increased concentration of this enzyme in the proximal nephron may facilitate the degradation of prostaglandins delivered to the proximal tubule by glomerular filtration.6
FIGURE 32.1 Synthetic and degradative pathways for the different types of eicosanoids. 15-PDG, 15 prostaglandin dehydrogenase; EETs, epoxyeicosatrienoic acids; HETE, hydroxyeicosatetraenoic acid; TXA2, thromboxane A2.
TABLE 32.1 Compartmentalization and Function of Renal Prostaglandins
Site
Eicosanoid
Action
Arterioles
PGI2, PGE2
Vasodilation
Glomeruli
PGI2 > PGE2 (human)
Maintain GFR
PGE2 >PGI2 (rat)
Vasoconstriction
TXA2
Tubules
PGE2, PGF2α
Enhance NaCl and water excretion
Interstitial cells
PGE2
Enhance NaCl and water excretion, influences regional blood flow
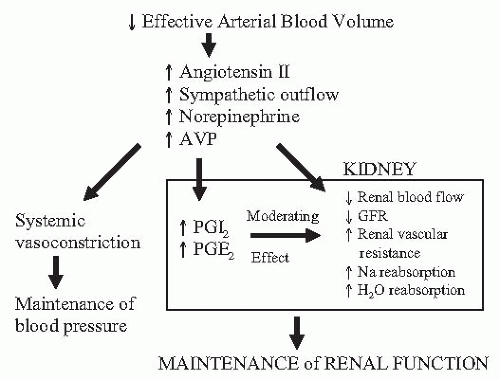
Under baseline euvolemic conditions, prostaglandin synthesis is negligible, and as a result, these compounds play little to no role in the minute-to-minute maintenance of renal function. However, a major role occurs in the setting of a systemic or intrarenal circulatory disturbance. This interaction is best illustrated when examining the renal function under conditions of volume depletion (Fig. 32.2). In this setting, renal blood flow is decreased while sodium reabsorption, renin release, and urinary concentrating ability are increased. To a large extent, these findings are mediated by the effects of increased circulating levels of angiotensin II (AII), arginine vasopressin (AVP), and catecholamines. At the same time, these hormones stimulate the synthesis of renal prostaglandins, which in turn act to dilate the renal vasculature, inhibit salt and water reabsorption, and further stimulate renin release. Prostaglandin release under these conditions serves to dampen and counterbalance the physiologic effects of the hormones that elicit their production. As a result, renal function is maintained near normal levels despite the systemic circulation being clamped down. Predictably, the inhibition of prostaglandin synthesis will lead to unopposed activity of these hormonal systems, resulting in exaggerated renal vasoconstriction and magnified antinatriuretic and antidiuretic effects. In fact, many of the renal syndromes that are associated with the use of NSAIDs can be explained by the predictions of this model.
FIGURE 32.2 In the setting of absolute or effective volume depletion, a number of effectors are activated that serve to defend the circulation and, at the same time, stimulate the synthesis of renal prostaglandins. In turn, renal prostaglandins function to moderate the effects of these hormonal systems such that renal function is maintained in the setting of systemic vasoconstriction. AVP, arginine vasopressin; GFR, glomerular filtration rate.
EEXPRESSION AND REGULATION OF CYCLOOXYGENASE-1 AND -2 IN THE KIDNEY
Aspirin and other NSAIDs exert their prostaglandin-inhibitory effects by inhibiting the COX enzyme. The COX enzyme exists as two isoforms termed COX-1 and COX-2. These enzymes are encoded by two different genes and differ significantly in their regulation. The COX-1 enzyme is constitutively expressed in most tissues and is responsible for producing prostaglandins involved in maintaining normal tissue homeostasis. The COX-2 enzyme is principally an inducible enzyme rapidly upregulated in response to a variety of stimuli such as growth factors and cytokines typically found in the setting of inflammation.3 With the discovery of COX-2, a great deal of effort was put forth to develop compounds to selectively block the activity of this isoform without affecting the activity of COX-1. The availability of a COX-2-specific inhibitor would provide a therapeutic tool to inhibit the synthesis of arachidonic acid metabolites at sites of inflammation and yet leave unperturbed COX-1-derived prostanoids involved in normal homeostasis. In this manner the analgesic, anti-inflammatory, and antipyretic effects of an NSAID could be obtained with minimal to no side effects. Although the initial experience with specific COX-2 inhibitors has been associated with a reduction in gastrointestinal complications, this paradigm is not applicable to the kidney.
COX-1 and COX-2 are both constitutively expressed in the kidney. COX-1 is localized to mesangial cells, arteriolar endothelial cells, parietal epithelial cells of the Bowman capsule, and throughout the cortical and medullary collecting duct.7 COX-2 is primarily expressed in the macula densa and the adjacent cells in the cortical thick ascending limb with lesser amounts in the podocytes and the arteriolar smooth muscle cells.8,9 COX-2 is also abundantly expressed in interstitial cells in the inner medulla and the papilla.
The expression of COX-2 in different regions of the kidney varies in response to alterations in intravascular volume. This variation is particularly evident in the macula densa, where studies show COX-2 plays an important stimulatory role in the release of renin via the tubuloglomerular feedback mechanism. Under conditions of low renal perfusion when the chloride concentration at the level of the macula densa is low, renin release is inhibited by a COX-2 selective inhibitor but unaffected by a COX-1 inhibitor.10 In genetically engineered mice lacking COX-2, there is a failure of renin release in response to a low salt diet, whereas renin release is intact in animals lacking COX-1.11,12
Stimulation of renin with the subsequent formation of angiotensin II is part of a feedback loop because angiotensin II exerts an inhibitory effect on COX-2 synthesis in the macula densa via the angiotensin type 1 (AT1) receptor.13 In contrast to effects at the macula densa angiotensin II upregulates COX-2 and prostaglandin synthesis in vascular smooth muscle cells and mesangial cells.14 This latter effect provides a mechanism for COX-2 to both facilitate the tubuloglomerular feedback response to low salt delivery to the macula densa by increasing angiotensin II levels and preserve the glomerular filtration rate through the generation of vasodilatory prostaglandins to antagonize the vasoconstrictive effect of angiotensin II.15
The expression of COX-2 is also responsive to changes in volume. COX-2 expression decreases with salt depletion and increases with a high salt diet and dehydration.9 COX-2-derived prostaglandins may play an important role in facilitating a natriuretic response to salt loading and may help protect against volume overload. The increase in COX-2 in response to dehydration is thought to provide a cytoprotective effect in the setting of hypertonic stress.16,17 Treatment of water-deprived animals with a selective COX-2 inhibitor is associated with apoptotic patches of renal medullary interstitial cells. By contrast, no such changes are seen in animals treated with the inhibitor alone or in animals undergoing water deprivation without pharmacologic treatment.
In summary, COX-2 is constitutively expressed in the kidney and is highly regulated in response to physiologic perturbations in intravascular volume. The majority of experimental and clinical studies to date suggest that the specific COX-2 inhibitors may not offer any distinct advantage over traditional NSAIDs with regard to renal toxicity. In fact, most of the renal syndromes that have been linked to nonselective COX inhibitors have now been described with the selective COX-2 inhibitors. The only exception is the development of chronic kidney disease and papillary necrosis. The failure to link COX-2 inhibitors use to these complications is not surprising because these agents have only been available for clinical use for a relatively short time. As with traditional NSAIDs, the COX-2 inhibitors need to be used cautiously and require close monitoring of renal function in patients at high risk for adverse renal outcomes.
EFFECTS OF PROSTAGLANDINS ON THE RENAL CIRCULATION
Prostaglandins primarily exert a vasodilatory effect on the renal vasculature. This vasodilatory effect alters the renal circulation in two major ways. First, these compounds influence the distribution of renal blood flow to different regions of the kidney. Prostaglandin stimulation results in a preferential increase in blood flow to the more juxtamedullary nephrons.18,19 By contrast, the inhibition of prostaglandin synthesis results in a selective reduction of flow to the inner cortical nephrons while flow remains well preserved in the outer cortex.20 Second, prostaglandins exert a vasoregulatory effect on the renal microcirculation to include the interlobular, afferent, and efferent arterioles as well as the glomerular mesangium. In isolated renal arterioles, both PGE2 and PGI2 attenuate AII-induced and norepinephrine-induced afferent arteriolar vasoconstriction. On the efferent side of the circulation, PGI2 similarly antagonizes AII-induced and norepinephrine-induced vasoconstriction, but PGE2 is without effect.21 In addition to local production, vascular reactivity of the efferent arteriole appears to be influenced by prostaglandins produced in the upstream glomerulus. In this regard, Arima and associates22 find that the orthograde infusion of AII (afferent arteriole-glomerulus-efferent arteriole) results in less vasoconstriction of the efferent arteriole as compared to when infused in a retrograde fashion (efferent arterioleglomerulus-afferent arteriole). Pretreatment with indomethacin markedly increases the vasoconstrictive effect during an orthograde infusion but is without effect during the retrograde infusion.
Prostaglandins have also been shown to attenuate mesangial cell contraction induced by AII, endothelin, AVP, and platelet-activating factor.23,24 Contraction of these cells will normally cause a decrease in the total glomerular capillary surface area and result in a fall in the GFR. Mesangial cell synthesis and the release of PGI2 in humans and PGE2 in rats dampen the constrictor effects of these hormones such that the glomerular capillary surface area is maintained, thereby minimizing any fall in GFR. Thus, in the setting of enhanced hormonal constrictor activity, prostaglandins play a major role in maintaining glomerular hemodynamics by exerting a vasodilatory effect at the level of the afferent and efferent arteriole as well as within the glomerular mesangium.
RENAL SYNDROMES ASSOCIATED WITH NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
Vasomotor-Induced Acute Renal Failure
Prostaglandins appear to play a negligible role in the maintenance of renal function under normal circumstances. This conclusion is based on studies in both experimental animals as well as humans. In conscious, sodium-replete dogs and rats, the inhibition of renal prostaglandin synthesis with a variety of NSAIDs does not alter baseline renal blood flow or GFR.25,26 Similarly, renal hemodynamics are unaffected in healthy humans after both the short-term27,28 and the long-term administration of aspirin.29 In related studies, the administration of indomethacin to healthy volunteers was also found to produce no change in renal hemodynamics.30
A sharply different effect of COX inhibition is observed when systemic hemodynamics are compromised. Under conditions of circulatory distress, renal blood flow represents a balance between vasoconstrictor influences on the one hand and vasodilatory prostaglandins on the other. Predictably, the administration of NSAIDs in this setting will shift this balance toward unopposed vasoconstriction and will potentially result in a precipitous decline in renal function.
This interplay between vasoconstrictive effectors and vasodilatory prostaglandins is particularly well illustrated in a series of studies using a model of hemorrhage in dogs.31,32 In animals subjected to a hemorrhage, prostaglandin synthesis inhibition was associated with a marked reduction in renal blood flow as compared to prostaglandin-intact dogs. This renal ischemic response was found to be partly reversed after the infusion of an AII antagonist or after renal denervation. When renal denervation was combined with the AII antagonist, renal blood flow was restored to values comparable to that in the nonprostaglandin-inhibited animals. These findings illustrate the pivotal role that prostaglandins play in opposing the renal ischemic effects of AII and renal nerves.
The modulating effect of vasodilatory prostaglandins on renal hemodynamics can be expected to roughly parallel the extent to which vasoconstrictor effectors are activated. In turn, the activity of these effectors will reflect the degree of circulatory distress. With only mild perturbations in the circulation, one can begin to detect a discernible effect of prostaglandins on renal blood flow. For example, unlike subjects ingesting an ad lib sodium diet, normal subjects placed on a salt-restricted diet will demonstrate a modest fall in creatinine clearance and renal blood flow following the administration of aspirin or indomethacin.33,34
Diuretic therapy is a common clinical situation where NSAIDs may exert a deleterious effect on renal function in otherwise healthy subjects.35 Like sodium restriction, diuretics increase the dependence of renal blood flow and GFR on vasodilatory prostaglandins and potentiate the deleterious effects of prostaglandin inhibition with COX inhibitors. The degree to which renal function is disturbed, however, appears to vary depending on which diuretic-NSAID combination is used. In this regard, Favre and colleagues35 found that the combination of triamterene and indomethacin given to healthy subjects results in a marked decline in creatinine clearance. By contrast, only a mild decrease in creatinine clearance is found when indomethacin is given in combination with furosemide, hydrochlorothiazide, or spironolactone. Interestingly, triamterene is the only diuretic associated with a marked increase in urinary prostaglandin secretion. Although there is little evidence to suggest that the renal failure patients in this study were volume-depleted, it appears that triamterene, by some unknown mechanism, renders the renal circulation critically dependent on vasodilatory prostaglandins. As a result, triamterene in combination with an NSAID should only be used with extreme caution.
As alterations in the circulation become more pronounced, rendering the renal circulation more dependent on vasodilatory prostaglandins, COX inhibition can be expected to result in more profound changes in renal hemodynamics. In congestive heart failure, a decrease in effective arterial circulatory volume is the proximate cause for activation of neurohumoral vasoconstrictor forces that participate in the maintenance of systemic arterial pressure and result in increased total peripheral vascular resistance. Important to note, the rise in renal vascular resistance is less than that seen in the periphery. Vasodilatory prostaglandins function in a counterregulatory role, attenuating the fall in renal blood flow and GFR that would otherwise occur if vasoconstrictor forces were left unopposed.36
Cirrhosis is another clinical condition in which the integrity of the renal circulation can become critically dependent on vasodilatory renal prostaglandins. Cirrhotic patients with a low urinary sodium concentration tend to be the most susceptible to develop acute decrements in renal function following the administration of NSAIDs.37 These patients have a more marked decrease in effective arterial circulatory volume, primarily due to splanchnic vasodilation, which in turn leads to higher levels of circulating catechols, AII, and AVP.38,39 As a result, the renal circulation in this subset of patients is more critically dependent on the effect of vasodilatory prostaglandins. As seen in patients with congestive heart failure, these patients have high urinary concentrations of PGE2, which decline in parallel with the fall in GFR.37
Renal prostaglandins may play an important role in the maintenance of renal hemodynamics in nephrotic syndrome. GFR and filtration fraction are moderately decreased in most patients with the nephrotic syndrome.40,41 Micropuncture studies in an experimental model of the nephrotic syndrome have indicated that the relative preservation of renal plasma flow may serve an important role in attenuating the fall in GFR that would otherwise occur due to a reduction in the ultrafiltration coefficient.42 In this setting, locally produced vasodilatory prostaglandins may serve to reduce afferent arteriolar resistance, thereby increasing renal plasma flow and increasing filtration pressure.43,44 The administration of NSAIDs in this setting would lead to increased afferent arteriolar tone. The resulting fall in renal plasma flow and filtration pressure combined with the already decreased ultrafiltration coefficient would result in a dramatic fall in GFR.43 Indeed, the administration of prostaglandin synthesis inhibitors to nephrotic subjects is commonly associated with a fall in GFR and may precipitate acute renal failure in some patients.45 Other settings in which there is an increased vasoconstrictive input focused on the kidney rendering it particularly vulnerable to the deleterious effects of NSAIDs include endotoxic shock46 and anesthesia.47
Risk factors for the development of NSAID-induced acute kidney injury are not necessarily confined to conditions characterized by decreases in absolute or effective arterial circulatory volume (Table 32.2). One such example is the presence of underlying chronic kidney disease. In this setting, increased vasodilatory prostaglandins are thought to play an adaptive role in minimizing the decline in global renal function by increasing GFR in surviving nephrons through increased renal blood flow. The signal for increased prostaglandin production is generally not a disturbance in the systemic circulation leading to increased circulating levels of AII and catecholamines but rather intrarenal mechanisms leading to the generation of vasoactive compounds within the glomerular microcirculation.48
TABLE 32.2 Risk Factors for NSAID-Induced Acute Vasomotor Renal Failure
Decreased EABV
Normal or ↑ EABV
Congestive heart failure
Chronic renal failure
Cirrhosis
Glomerulonephritis
Nephrotic syndrome
Elderly
Sepsis
Contrast-induced nephropathy
Hemorrhage
Obstructive uropathy
Diuretic therapy
Cyclosporin A
Postoperative patients with “third space” fluid
Volume depletion/hypotension
EABV, effective arterial blood volume.
TABLE 32.3 Predisposing Factors for NSAID-Induced Nephrotoxicity in the Elderly
Age-related changes in renal function
↓ in glomerular filtration rate
↓ in renal blood flow
↑ in renal vascular resistance
Age-related changes in pharmacokinetics
↑ free drug concentration
– Hypoalbuminemia
– Retained metabolites
↓ total body water
↓ hepatic metabolism resulting in longer drug half-life
Increasing age is a risk factor for the development of nephrotoxicity when using NSAIDs.49,50 This susceptibility, in part, may be related to changes in kidney function that normally accompany the aging process (Table 32.3).51 Aging is associated with a progressive decline in the GFR and total renal blood flow. In addition, there is an increase in renal vascular resistance. Important to note, the renal vasculature becomes less responsive to vasodilators, whereas the response to vasoconstrictors remains intact. In an analysis of 1,908 patients treated with ibuprofen, renal impairment was found to occur in 343 (18%) patients.49 The two most important risk factors identified for the development of toxicity is an age greater than 65 years and preexisting renal insufficiency. In a prospective study of 114 older patients (mean age 87 years) started on NSAID therapy, a greater than 50% increase in the serum urea nitrogen concentration was found in 15 (13%) patients.50 In this study, the concurrent use of a loop diuretic and large doses of NSAIDs were found to be predictive of those who developed significant azotemia.
In addition to age-related changes in kidney function, age-related changes in the pharmacokinetics of NSAIDs may also make this population more susceptible to renal toxicity.52,53 Older patients, particularly those with chronic illness, often have lower albumin levels, which reduce the protein binding of the drugs and result in higher free-drug concentrations. This binding of the parent compound to circulating albumin is further impaired by retained metabolites, which accumulate as a result of the normal age-related impairment in renal function. Increased drug levels also occur as a result of the age-related decrease in total body water. Finally, decreased hepatic metabolism, which is often present in older adults, contributes to a longer half-life of the parent compound and can result in unexpectedly high drug levels.
Other conditions in which effective arterial circulatory volume is normal or increased and yet renal function is critically dependent on increased synthesis of prostaglandins include immune mediated glomerular injury, urinary obstruction,54 radiocontrast-induced injury,55 and the administration of calcineurin inhibitors.56 In these conditions, the increased production of vasodilatory prostaglandins has been shown to counterbalance the effects of intrarenally generated vasoconstrictors such as thromboxane, leukotrienes, platelet-activating factor, and endothelin. The administration of NSAIDs in each of these settings can be expected to result in an exaggerated fall in renal function.
NSAID-induced acute kidney injury is most commonly an oliguric form of renal failure that begins within several days after the initiation of the drug (Table 32.4). The urinalysis is unremarkable in the majority of cases. Unlike other causes of acute kidney injury, the fractional excretion of sodium is often less than 1%. This low fractional excretion of sodium reflects the underlying hemodynamic nature of the renal failure. Hyperkalemia out of proportion to the decrement in renal function is also a typical feature of this lesion. If recognized early, the renal failure is reversible with the discontinuation of the NSAID. As a result, dialysis is usually not required.
Glomerular and Interstitial Disease
The use of NSAIDs can be associated with the development of a distinct syndrome characterized by the development of interstitial nephritis and nephrotic range proteinuria. The incidence of this lesion is unknown but is thought to be rare. One estimate for fenoprofen-induced interstitial nephritis was 1 case per 5,300 patient-years of treatment.57 Although virtually all NSAIDs have been reported to cause this syndrome, the vast majority of cases have been reported in association with the use of propionic acid derivatives (fenoprofen, ibuprofen, and naproxen). Of these, fenoprofen has been implicated in greater than 60% of cases.58 Interstitial nephritis with and without nephrotic syndrome has also been reported with the COX-2 inhibitors, rofecoxib and celecoxib.59,60,61,62,63
Unlike hemodynamically mediated ARF, there are no clear-cut risk factors that serve to identify those at risk for the development of this syndrome. The mean age of patients is 65 years.58 The presence of an underlying renal disease prior to the exposure of the NSAID has been notably absent. This syndrome has generally been referred to as an example of acute interstitial nephritis. There are, however, a number of features that distinguish this form of interstitial renal disease from that observed with other pharmacologic agents (Table 32.5).58 First, the average duration of exposure prior to the onset of the disease is typically measured in months and can be as long as a year. By contrast, allergic interstitial nephritis due to other drugs usually presents within several days to weeks after exposure to the drug. Second, nephrotic range proteinuria is found in >90% of cases of NSAID-induced interstitial disease, a degree of proteinuria that is distinctly uncommon in acute allergic interstitial nephritis due to other drugs. Third, symptoms of hypersensitivity that are commonly seen in acute allergic interstitial nephritis such as rash, fever, arthralgias, or peripheral eosinophilia are uncommon in NSAID-associated disease. Fourth, the vast majority of cases associated with NSAIDs have been reported in older patients. On the other hand, allergic interstitial nephritis is seen in all age groups.
TABLE 32.4 Clinical Features of NSAID-Induced Vasomotor Renal Failure
• Oliguria
• Usually occurs within a few days of beginning medicine
• Hyperkalemia out of proportion to renal failure
• Low fractional excretion of Na
• Usually does not require dialysis
• Usually reversible
Only gold members can continue reading. Log In or Register to continue
May 29, 2016 | Posted by drzezo in NEPHROLOGY | Comments Off on Nephrotoxicity of Nonsteroidal Anti-inflammatory Agents, Analgesics, and Inhibitors of the Renin-Angiotensin System