Miscellaneous Urological Diseases of the Kidney
Miscellaneous Urological Diseases of the Kidney
Simple and complex renal cysts
Simple renal cysts: do not communicate with any part of the nephron or the renal pelvis. They are mainly confined to the renal cortex, are filled with clear fluid, and contain a membrane composed of a single layer of flattened or cuboidal epithelium. They can be single or multiple, ranging from a few millimetres to several centimetres in diameter. They can be unilateral or bilateral and often affect the lower pole of the kidney.
Parapelvic cysts: describe simple parenchymal cysts located adjacent to the renal pelvis or hilum.
Prevalence
Increases with age, the precise prevalence depending on the method of diagnosis. On
CT, 20% of adults have renal cysts by age 40y and 33% by the age of 60.
1 At post-mortem, 50% of subjects aged >50y have simple cysts. Cysts do not usually increase in size with age, but may increase in number. Males and females are affected equally.
Aetiology
Both congenital and acquired causes have been suggested. Chronic dialysis is associated with the formation of new simple cysts.
Presentation
Simple cysts are most commonly diagnosed as an incidental finding following a renal ultrasound scan (
USS) or
CT performed for other purposes. The majority are asymptomatic; however, very large cysts may present as an abdominal mass or cause dull flank or back pain. Acute severe loin pain may follow bleeding into a cyst (causing sudden distension of the wall). Rupture (spontaneous or following renal trauma) is rare. Rupture into the pelvicalyceal system can produce haematuria. Infected cysts (rare) present with flank pain and fever. Very occasionally, large cysts can cause obstruction and hydronephrosis.
Investigation
Renal USS
Simple cysts are round or spherical, have a smooth and distinct outline, and are ‘anechoic’ (no echoes within the cyst,
i.e. sound waves are transmitted through the cyst).
USS using microbubble contrast agents can improve diagnostic accuracy. Evidence of calcification, septation, irregular margins or clusters of cysts requires further investigation (renal triphasic
CT). In the absence of these features, no further investigation is required.
CT
Simple cysts are seen as round, smooth-walled lesions with homogenous fluid in the cavity (with a typical density of-10 to +20 Hounsfield units) and with no enhancement after contrast (enhancement implies that it contains vascular tissue or communicates with the collecting system,
i.e. that it is not a simple cyst). Hyperdense cysts have a density of +20-90 Hounsfield units, do not enhance with contrast media, and are <3cm in diameter.
Biopsy
Image-guided cyst aspiration or biopsy can be used to help diagnose indeterminate cysts and prevent unnecessary surgery.
1 Laucks SP Jr, McLachlan MS (1981) Aging and simple cysts of the kidney. Br J Radiol 54:12?.
2 Warren KS, McFarlane J (2005) The Bosniak classification of renal cystic masses. BJU Int 95: 939?2.
Calyceal diverticulum
A calyceal diverticulum is a spherical outpouching of the renal collecting system (specifically from a calyx) which protrudes into the corticome-dullary region of the kidney. It communicates with the renal calyx via a narrow neck or channel. It is lined by a transitional cell epithelium and is covered by a thin layer of renal cortex. They range in size from only a few millimetres to many centimetres in size.
Aetiology
The exact aetiology of calyceal diverticula is unknown. Some may be congenital. Acquired calyceal diverticula can develop after obstruction of a calyceal infundibulum or following blunt renal trauma.
Presentation
They are usually asymptomatic and are discovered incidentally on an
IVU, most commonly seen in upper pole calyces. Symptoms may result from the development of a stone or infection within the diverticulum, presumably caused by urinary stasis.
Investigation
On
IVU, a calyceal diverticulum appears as a rounded collection of contrast medium next to a papilla, although often, the connecting channel is too narrow to be clearly seen. They can be identified on
CT,
MRI, and
USS; however, the distinction between a renal cyst and an obstructed calyx may be difficult on unenhanced images.
Medullary sponge kidney (MSK)
Prevalence
Difficult to know as it may be asymptomatic (diagnosed on an
IVU performed for other reason or at post-mortem). Estimated to affect between 1 in 5000 to 1 in 20 000 people in the general population; 1 in 200 in those undergoing
IVU (a select population). In 75% of cases, both kidneys are affected.
Pathology
The renal medulla resembles a sponge in cross section due to dilated collecting ducts in the renal papillae and the development of numerous small cysts. This is associated with urinary stasis and the formation of small calculi within the cysts. Some report a familial inheritance. It can be associated with other congenital or inherited disorders, including hemihypertrophy and Beckwith-Wiedemann syndrome
*.
Presentation
The majority of patients are asymptomatic. When symptoms do occur, they include ureteric colic, renal stone disease (calcium oxalate ± calcium phosphate),
UTI, and haematuria (microscopic or macroscopic). Up to 50% have hypercalciuria due to renal calcium leak or increased gastrointestinal calcium absorption. Renal function is normal unless obstruction occurs (secondary to renal pelvis or ureteric stones).
Investigation
Midstream urine: dipstick ± culture. Check for
UTI and treat according to sensitivities.
Biochemistry: 24h urinary calcium may be elevated (hypercalciuria). Detection of hypercalciuria requires further investigation to exclude other causes (
i.e. raised serum parathyroid hormone (
PTH) levels indicate hyperparathyroidism).
Imaging: IVU is the principle method for diagnosing
MSK, although
CT and
USS may also be used. The characteristic radiological features of
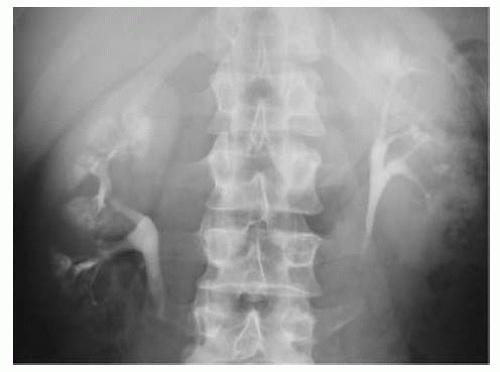
MSK, as seen on
IVU, are enlarged kidneys associated with dilatation of the distal portion of the collecting ducts, along with numerous associated cysts and diverticula (the dilated ducts are said to give the appearance of ‘bristles on a brush’). The collecting ducts may become filled with calcifications, giving an appearance described as a ‘bouquet of flowers’ or ‘bunches of grapes’ (
Fig. 8.1).
* Beckwith-Wiedemann syndrome: a growth disorder characterized by macroglossia, macroso-mia, visceromegaly, Wilm’s tumour, neuroblastoma, omphalocele, and renal anomalies.
Acquired renal cystic disease (ARCD)
A cystic degenerative disease of the kidney with ≥5 cysts visualized on
CT scan. By definition, this is an acquired condition, as opposed to
ADPKD which is inherited (in an autosomal dominant fashion). It is predominantly associated with chronic and end-stage renal failure and as such, is commonly found in patients undergoing haemodialysis or peritoneal dialysis. Over one third of patients develop
ARCD after 3y of dialysis. Clinically important because it may cause pain and haematuria and is associated with the development of benign and malignant renal tumours. The male to female ratio is 3:1.
Pathology
Usually multiple, bilateral cysts found mainly within the cortex of small, contracted kidneys. Cysts vary in size (average 0.5-1cm) and are filled with a clear fluid which may contain oxalate crystals. They usually have cuboidal or columnar epithelial linings and are in continuity with renal tubules (and, therefore, cannot be defined as simple cysts). Atypical cysts have a hyperplastic lining of epithelial cells, which may represent a precursor for tumour formation. Renal transplantation can cause regression of cysts in the native kidneys.
Aetiology
The exact pathogenesis is unknown, but several theories have been proposed. Obstruction or ischaemia of renal tubules may induce cyst formation. Renal failure may predispose to the accumulation of toxic endogenous substances or metabolites, alter the release of growth factors, and result in changes in sex steroid production or cause cell proliferation (secondary to immunosuppressive effects) which result in cyst formation.
Presentation
Flank pain;
UTI; visible haematuria; renal colic (stone disease); hypertension.
Investigation
This depends on the presenting symptoms.
For suspected UTI: culture urine.
For haematuria: urine cytology, flexible cystoscopy, and renal
USS. On
USS, the kidneys are small and hyperechoic, with multiple cysts of varying size, many of which show calcification. If the nature of the cysts cannot be determined with certainty on
USS, arrange a renal
CT.
Autosomal dominant polycystic kidney disease (ADPKD)
Epidemiology
Incidence is 0.1-0.5%; 95% are bilateral.
ADPKD can affect children and adults, although symptoms usually occur between ages 30-50y.
ADPKD accounts for 10% of all renal failure (which usually manifests at >40y old).
Pathology
The kidneys reach an enormous size due to multiple fluid-filled cysts and can easily be palpated on abdominal examination. Expansion of the cysts results in ischaemic atrophy of the surrounding renal parenchyma and obstruction of normal renal tubules. End-stage renal failure occurs at around age 50y.
Associated disorders
Ten to thirty percent incidence of Circle of Willis berry aneurysms (associated with subarachnoid haemorrhage), cysts of the liver (33%), pancreas (10%), spleen (<5%) and seminal vesicles, mitral valve prolapse; aortic root dilatation, aortic aneurysms, and diverticular disease. Of note, the incidence of renal adenoma is ˜20%; however, the risk of
RCC is the same as the general population.
Aetiology
Two genes have been identified in
ADPKD. The PKD1 gene is localized on the short arm of chromosome 16 (16p13.3) and accounts for 85% of cases. The PKD2 gene is on the long arm of chromosome 4 (4q21) and causes 15% of cases. A third gene, PKD3, is also implicated. Pathogenesis theories include intrinsic basement membrane abnormalities, tubular epithelial hyperplasia (causing tubular obstruction and basement membrane weakness), and alterations in the supportive extracellular matrix due to defective proteins, all of which may cause cyst formation.
Get Clinical Tree app for offline access