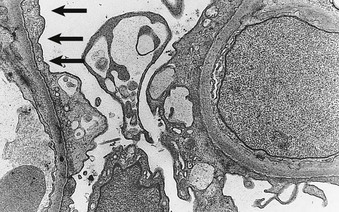
Philip D. Mason, Peter F. Hoyer Minimal change disease (MCD) is the cause of nephrotic syndrome in about 90% of children younger than 10 years, about 50% to 70% of older children, and 10% to 15% of adults. MCD is defined by the absence of histologic glomerular abnormalities, other than ultrastructural evidence of fusion of epithelial cell foot process, in a patient presenting with nephrotic syndrome who is typically corticosteroid responsive. Corticosteroid-sensitive nephrotic syndrome is the term used to describe the disease occurring in children with nephrotic syndrome who respond to corticosteroids but who have not had a renal biopsy to provide the histologic proof of MCD. The presence of nephrotic syndrome is important because similar histologic findings may be seen in patients with proteinuria in the absence of nephrotic syndrome. Such patients may have different conditions with different prognoses and requirements for management. Whereas MCD is classically associated with normal-appearing glomeruli and corticosteroid responsiveness, MCD appears to overlap with a variety of histologic variants that have a tendency to be less corticosteroid responsive. These conditions include focal segmental glomerulosclerosis (FSGS; see Chapter 18) and IgM nephropathy (see later discussion). It is possible that both MCD and FSGS have similar initial histologic appearances, but that FSGS is less corticosteroid responsive and thus develops secondary sclerosing lesions over time. Whether this represents a continuum of the same disease, in which some patients are corticosteroid sensitive and others are not, or whether these represent two distinct etiologies remains debatable. In children age 2 to 12 years, MCD is the most common cause of nephrotic syndrome. However, it is also relatively common in adults. Many patients have a history of allergy, including atopy, asthma, or eczema (Box 17-1). Most cases are idiopathic, but MCD has also been associated with certain cancers, especially Hodgkin disease, in which it may be the presenting symptom. MCD is further associated with certain medications, such as interferon alfa and nonsteroidal anti-inflammatory drugs (NSAIDs). The primary abnormality in MCD is a defect in the glomerular filtration barrier to protein. In normal glomeruli, the barrier to protein filtration is provided by the glomerular basement membrane (GBM) and the slit diaphragm that extends between the podocyte foot processes. Size barriers and charge selectivity of GBM exclude neutral molecules larger than 4 to 4.5 nm from filtration; albumin molecules smaller than this are excluded because they are anionic and are repelled by the negative charge on the epithelial cells and GBM. In MCD, FSGS, and other causes of nephrotic syndrome, the clearance of proteins becomes greatly enhanced, whereas the filtration of small molecules is actually reduced. Some authorities have attributed the reduction in small molecular clearance to the diffuse foot process fusion, which reduces overall slit diaphragm area and thereby may account for the reduction in the ultrafiltration coefficient (Kf, see Chapter 2) typically observed. In contrast, the albuminuria is thought to result from specific areas (“large pores”) where the protein escapes into the urine. Studies in nephrotic syndrome have implicated injury to the podocyte, and particularly the slit diaphragm, as the key factor leading to proteinuria. The importance of the slit diaphragm for nephrosis was first shown with the congenital nephrotic syndrome of the “Finnish type,” in which the primary defect is a mutation in nephrin, a key protein in the slit diaphragm. In particular, this nephrotic syndrome can histologically and clinically appear identical to MCD, except that it typically begins shortly after birth and is corticosteroid resistant (see Chapter 19). Since the discovery of the etiology of this nephrotic syndrome, numerous other genetic causes of nephrotic syndrome have been identified. All have mutations in genes involved in the development, structure, and function of podocytes (NPHS2, NPHS3, WT1, LAMB2, LMXI2, ACTN4, CD2AP, TRPC6, and INF2) and mitochondrial proteins (e.g., COQ2, PDSS2, GMS1). As evidence for involvement of a primary podocyte abnormality, MCD is associated with expression of CD80 (B7.1) on the podocyte, and shed CD80 can be found in the urine of patients with MCD.1 CD80 expression by podocytes is associated with shape change and development of proteinuria. Reduced levels of dystroglycans (adhesion molecules believed to anchor podocytes to GBM) have also been reported in MCD but not in FSGS, with normalization after corticosteroid treatment.2 Podocyte overexpression of angiopoetin-like-4 (ANGPTL4) has been shown to bind to the GBM, with loss of charge and associated with nephrotic-range proteinuria. Furthermore, supplementation with N-acetyl-d-mannosamine may improve proteinuria, suggesting that hyposialysated ANGPTL4 may be responsible for inducing proteinuria and may be a therapeutic target.3 Others have proposed that the permeability defect is caused by alterations in the GBM, particularly a loss of negative charge. In this case, podocyte changes could be secondary to the proteinuria. This would be consistent with observations of preserved foot processes in the early phase of some highly proteinuric states (e.g., recurrent FSGS after renal transplantation) and of podocyte foot process fusion in children with severe hypoalbuminemia, but not proteinuria, dying of kwashiorkor.4 Evidence also suggests that a circulating factor may be responsible for inducing the alteration in the capillary wall. Hemopexin, an acute-phase reactant extracted from human plasma, is capable of inducing proteinuria in rats,5 and in one small study, plasma hemopexin activity was increased in MCD patients in relapse, with evidence of an altered isoform.6 Evidence of a circulating factor in humans is also supported by the observation that proteinuria resolved within days after transplantation from a cadaveric donor with MCD.7 Evidence for a T cell abnormality includes the absence of immune deposits, the remarkable sensitivity of the disease to corticosteroids and cyclosporine, the relationship with Hodgkin disease, and the reports that active measles infection (which depresses cell-mediated immunity) can rapidly induce remission. Studies have shown that T regulatory cells are abnormal in MCD patients, and that generalized activation of T cells occurs during active disease. Some studies have also implicated interleukin-13 (IL-13) released from T cells in causing the proteinuria, because T cell production of IL-13 is elevated, and overexpression of IL-13 can induce nephrotic syndrome in rats.8 Interestingly, the mechanism involved the induction of CD80 in the podocyte. IL-13 is also a cytokine associated with allergic states, and MCD can be triggered by vaccination9 or exposure to an allergen in sensitive individuals. As a result, patients with identified food allergies have been managed with exclusion diets, with reported complete or partial remission and with relapse after reintroduction of the offending food. However, even if a relationship exists, the allergic events may merely trigger relapse, as may infections. B cell involvement was based on some patients’ response to rituximab, which binds to CD20 found on B cells. Recently, however, CD20 expression has been demonstrated on podocytes and may be upregulated in abnormal circumstances, and rituximab may also interact with podocyte sphingomyelin phosphodiesterase–like 3 protein.10 Bisphosphonate therapy may lead to proteinuria, possibly through interference with the podocyte cytoskeleton. Although steroids are first-line treatment for patients with MCD, the mechanism is unclear, and the optimal dose has never been formally tested. Previous studies describe an association between risk of relapse and suppression of the hypophyseal-adrenal axis.11 Recent studies report melanocortin-1 receptors on podocytes and, interestingly, a significant reduction of proteinuria with adrenocorticotropic hormone (ACTH) therapy.12 Most frequently presenting between 2 and 7 years of age, MCD affects 2 to 7 per 100,000 children per year, with a prevalence of 15 per 100,000. MCD is also an important cause of nephrotic syndrome in adults of all ages, although the incidence varies geographically. MCD is reported to be as low as 1 per million of the population in the United Kingdom and up to 27 per million in the United States. It is more common in South Asians and Native Americans but is much rarer in African Americans, in whom nephrotic syndrome is much more likely to be caused by FSGS and to be corticosteroid resistant. MCD is also relatively rare in developing countries, such as most countries in Africa and South America13–15 (Table 17-1). Boys are twice as likely to be affected as girls, but the gender incidence is comparable in adolescents and adults. Data also suggest that MCD has become less common in adults.16 Table 17-1 Frequency of various types of nephrotic syndrome. In annual incidence per 1 million population, in children and adults, from Europe, the United States, and Africa. MCD, Minimal change disease; FSGS, focal segmental glomerulosclerosis; MN, membranous nephropathy; MPGN, membranoproliferative glomerulosclerosis. * 35 with hepatitis B virus infection. (Data from references 13, 14, and 15.) Patients with MCD typically present with edema that develops during days to weeks, with fluid retention that often exceeds more than 3% of the body weight. Up to two thirds of initial presentations and relapses follow an infection, most often of the upper respiratory tract, but whether these are of causative significance is uncertain. The clinical signs and symptoms of MCD are the same as for nephrotic syndrome of any cause (see Chapter 15), although nephrotic syndrome is often of rapid onset, increasing the risk of hypovolemia, particularly in children. Ascites is common, particularly in children with MCD, who may present with abdominal pain, a symptom that may suggest peritonitis or herald hypovolemia. Pleural effusions may also occur but are less common. In rare patients, pericardial effusions may occur, but even more rarely cause significant complications. Pulmonary edema is uncommon, except after excessive treatment with albumin or with coexisting cardiac disease. Hepatomegaly is common in children but may be overlooked in the presence of ascites. The distribution of edema is gravitational, but facial puffiness is common, and genital swelling may be extremely uncomfortable, especially in men. Gross edema may predispose to ulceration and infection of dependent skin; striae often appear even without corticosteroids, and lacerations or needlestick punctures weep fluid profusely. Edema of the bowel wall may cause diarrhea, rarely with significant albumin loss from the gut. Other clinical features include white nails, sometimes in bands (Muehrcke lines) correlating with periods of clinical relapse (see Fig. 15-4). In adults, xanthomas may occasionally result in association with gross hyperlipidemia. Microscopic hematuria is rare in MCD. Although not typical in children, hypertension has been observed at presentation in 30% of 89 adults in one UK study.17 Higher-than-normal blood pressure has also been described in 14% to 21% of children, compared with appropriate age- and gender-matched blood pressure reference ranges. Hypertension usually resolves during remission, especially in children. Hypertension is sometimes associated with expansion of the intravascular volume but may paradoxically be related to hypovolemia and stimulation of the renin-angiotensin system (RAS). Before the introduction of corticosteroids, the morbidity and mortality of patients with MCD were high because of complications of nephrotic syndrome, particularly infection. Infection continues to be a serious problem, particularly in those presenting late.18 Six of 389 children with MCD described by the International Study of Kidney Disease in Children (ISKDC) in 1984 died of sepsis.19 Peritonitis remains a major cause of mortality in the developing world, mainly in children. Streptococcus pneumoniae, Haemophilus influenzae, and other encapsulated bacteria are implicated. Children with frequently relapsing nephrotic syndrome should be immunized against S. pneumoniae and H. influenzae during remission and given prophylactic oral penicillin in relapse.20 Peritonitis is rare in adults, who usually have protective antibodies against these bacteria, and prophylactic antibiotics are not indicated. The risk of thromboembolism is increased in MCD, as in all patients with nephrotic syndrome (see Chapter 15). Venous thromboembolism may occur in the lower extremities, renal veins, and other sites. Pulmonary embolism may be overlooked in children because of a lack of suspicion, even with pulmonary symptoms, and children may compensate better than adults. Nephrotic children occasionally have other catastrophic events, such as intracerebral venous or sinus venous thrombosis. Arterial thrombosis is a rare but feared complication, described in children almost exclusively, and may even result in loss of limbs. Renal function is generally preserved. Serum creatinine concentrations are usually low in children but can be slightly elevated in adults. Acute kidney injury is a complication particularly seen in adults. AKI may follow hypovolemia, which should be avoided especially during intensive diuretic treatment, but it may also rarely occur in volume-replete patients. Secondary MCD may mimic idiopathic MCD and may result from drugs or with cancer. The classic drugs associated with MCD are NSAIDs and particularly fenoprofen. This is an idiosyncratic reaction and is usually associated with chronic NSAID use that has occurred for several weeks or months. Unlike classic MCD, this syndrome is usually associated with massive nephrotic syndrome with impaired renal function, and renal biopsy shows MCD with features of an acute interstitial nephritis with T cell infiltration. Other causes of secondary MCD, such as the use of interferon alfa or interferon beta, or MCD observed in Hodgkin disease, may appear clinically identical to idiopathic MCD. Classically, MCD is associated with normal-appearing glomeruli by light microscopy and is negative for immunoglobulins and complement by immunofluorescence or other methodologies. Podocyte (epithelial cell) foot process effacement is observed with electron microscopy (Fig. 17-1) and is the only abnormality, but this is a nonspecific finding. The tubulointerstitium will show an absence of inflammation. Hyaline casts obstructing tubules, rare foam cells, and occasionally appearances consistent with acute tubular necrosis may be seen, especially if acute kidney injury is present at the time of biopsy. Mild mesangial hypercellularity is an infrequent finding in MCD patients (3% to 5%), and small amounts of mesangial IgG, complement C3, and occasionally IgA may be observed in patients whose clinical course is indistinguishable from classic MCD. The glomerular tip lesion describes structural segmental changes adjacent to the tubular pole of Bowman capsule, with protrusion into the tubular lumen. This lesion is observed more frequently in adults than children, but whether it is a variant of MCD or a type of FSGS remains controversial. Generally considered a benign lesion, it may also be rarely observed in membranoproliferative glomerulonephritis, IgA nephropathy, and renal allografts. The most important reason to recognize the glomerular tip lesion is to prevent a misdiagnosis of a proliferative glomerulonephritis. The presence of mesangial hypercellularity in MCD may correlate with increased resistance to corticosteroid therapy.21 Some consider mesangial hypercellularity to be an intermediate step in cases of evolution (progression) of MCD to FSGS (see Chapter 18). Some patients presenting with nephrotic syndrome have mesangial deposits of IgM, often with a minor degree of mesangial hypercellularity. Patients are more likely to have microscopic (and occasionally macroscopic) hematuria and are also less likely to respond to corticosteroids (50% versus 90% for minimal change nephrotic syndrome).22 Whether this represents a distinct entity or is part of the spectrum observed with MCD and FSGS remains uncertain (see also Chapter 18). Some patients who are considered to have MCD by renal biopsy may not respond to corticosteroids and on repeated biopsy are found to have FSGS. Others who initially are responsive to corticosteroids eventually become resistant and are discovered to have FSGS on a subsequent biopsy. This has led some to suggest that MCD and FSGS are part of a spectrum of the same disease process. However, the initial biopsies may have missed the sclerotic lesions of FSGS because they are focal. The clinical diagnosis of nephrotic syndrome is straightforward, with edema in the presence of heavy proteinuria, usually without microscopic hematuria on urine dipstick testing. Urine microscopy reveals hyaline casts and sometimes lipid casts. There is hypoalbuminemia (<2.5 g/dl) and nephrotic-range proteinuria (>3.5 g/24 h in adults or >1 g/m2/24 h [>40 mg/m2
Minimal Change Nephrotic Syndrome
Definition
Etiology and Pathogenesis
Epidemiology
Frequency of Types of Nephrotic Syndrome in Children and Adults
Histology
Children
Adults
Zimbabwe
Durban
MCD
76
20
9.2
14
FSGS
8
15
15.1
28
MN
7
40
15.1
41*
MPGN
4
7
33.6
9
Other
5
18
17.0
5

Clinical Manifestations
Pathology
Variants
IgM Nephropathy
Focal Segmental Glomerulosclerosis
Diagnosis and Differential Diagnosis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Minimal Change Nephrotic Syndrome
Chapter 17
Figure 17-1 Podocyte foot process fusion in minimal change disease. The epithelial cells (arrows) are completely effaced along the glomerular basement membranes. (Electron micrograph; magnification ×6000.) The normal appearance of epithelial cell foot processes is shown in Figures 1-6 and 1-7 (see Chapter 1).






