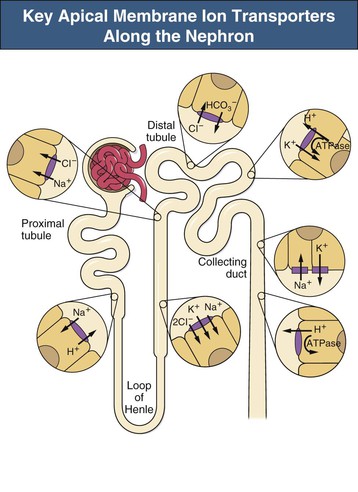
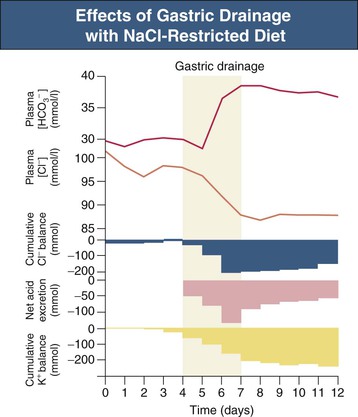
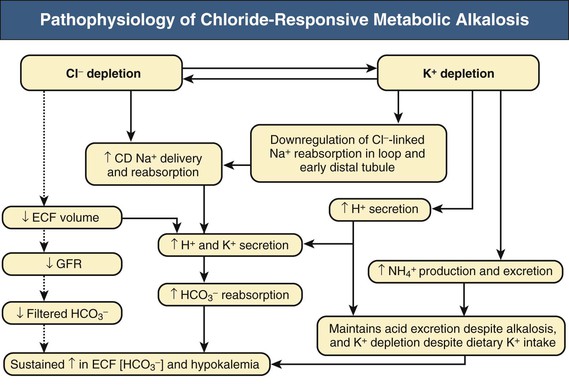
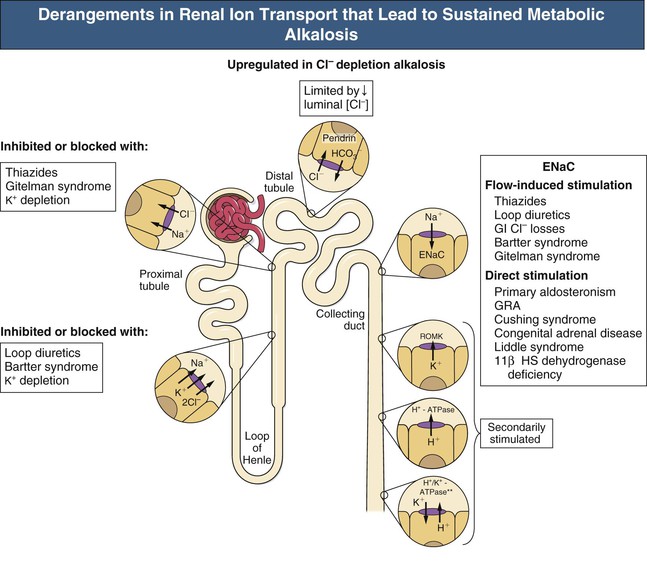
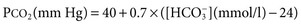F. John Gennari Metabolic alkalosis is caused by retention of excess alkali and is defined by a venous total carbon dioxide concentration ([total CO2]) greater than 30 mmol/l or an arterial bicarbonate concentration ([HCO3−]) greater than 28 mmol/l. The increase in pH that results from the elevation in [HCO3−] induces hypoventilation, producing a secondary increase in arterial CO2 tension (Paco2). Thus, metabolic alkalosis is characterized by coexisting elevations in serum [HCO3−], arterial pH, and Paco2. Because the kidney normally responds to an increase in [HCO3−] by rapidly excreting excess alkali, sustained metabolic alkalosis occurs only when some additional factor impairs bicarbonate excretion. This chapter reviews the normal process of bicarbonate reabsorption by the kidney and how it is impaired in metabolic alkalosis, the various clinical settings in which this disorder occurs, and its diagnosis and management. Bicarbonate ions are freely filtered across the glomerulus and under normal conditions must be completely recaptured from the tubule urine to conserve body alkali stores. In addition, acid excretion must occur to regenerate any HCO3− consumed in buffering of endogenously produced acids. Both tasks are accomplished by secretion of hydrogen ions (H+) into the renal tubules.1 Bicarbonate ions are recaptured when secreted H+ combines with filtered HCO3− to produce CO2 and water, removing HCO3− from the urine. Acid excretion occurs in the collecting duct when secreted H+ combines with filtered phosphate, converting HPO42− to H2PO4−, or with ammonia (NH3) to form ammonium (NH4+), and these ions are excreted. Whenever excess alkali is ingested or administered, HCO3− is secreted into the tubule urine as well as being filtered, and acid excretion decreases, all changes that result in rapid bicarbonate excretion. Figure 13-1 illustrates the key epithelial transporters and channels that participate in HCO3− reabsorption and secretion. In the proximal tubule, where most of the filtered HCO3− is reabsorbed, H+ is secreted into the tubule urine both by a Na+-linked transporter (Na+-H+ exchanger, NHE3) and an H+-ATPase (not shown in figure). In the thick ascending limb of Henle loop (TAL), H+ is secreted via NHE3, and in the collecting duct, H+ secretion is accomplished primarily by an apical membrane H+-ATPase. The activity of this collecting duct proton transporter is regulated by aldosterone and by the rate of Na+ delivery to and reabsorption at this site in the nephron. When body K+ stores are low, an apical membrane H+,K+-ATPase is activated in the cortical collecting duct, further promoting H+ secretion linked to K+ reabsorption.2 Excretion of excess bicarbonate is facilitated by secretion of HCO3− into the tubule urine in the cortical collecting duct through an apical membrane Cl−-HCO3− exchanger (pendrin).3 This transporter is activated by alkalemia and requires Cl− reabsorption in exchange for secreted HCO3−. The HCO3−secreted into the urine by this transporter can be recaptured again by H+ secretion further along in the collecting duct, so that excretion of excess alkali requires both stimulation of the Cl−-HCO3− exchanger and suppression of the normally active collecting duct H+-ATPase. With the exception of alkali administration in the setting of severe impairment of kidney function (GFR <30 ml/min), sustained metabolic alkalosis is always the result of dysregulation of the transporters that control bicarbonate reabsorption and acid excretion by the kidney. A key feature of this dysregulation is abnormal stimulation of collecting duct ion transport4 (Box 13-1). This stimulation usually is secondary to abnormalities in Na+ and Cl− reabsorption that occur before the urine reaches the collecting duct. More rarely, metabolic alkalosis is the result of primary stimulation of collecting duct ion transport resulting from abnormal signaling or genetic abnormalities. The most common clinical presentation of metabolic alkalosis is generated by Cl− depletion. Although the term contraction alkalosis is sometimes used as a synonym for Cl− depletion alkalosis, this phrase is confusing because it implies that volume contraction is responsible for the disorder. The term refers specifically to the increase in serum [HCO3−] that follows only one type of extracellular fluid (ECF) volume contraction, that caused by selective Cl− losses. Moreover, volume contraction is neither necessary nor instrumental in inducing this change in serum [HCO3−].5 Selective Cl− depletion, induced by vomiting or nasogastric suction, increases serum [HCO3−].6 The degree of alkalosis generated is greater when H+ loss also occurs, as shown in the example in Figure 13-2, but it may occur even when H+ loss is minimized by administration of a proton pump inhibitor. In either setting, maintenance of the metabolic alkalosis depends on sustained depletion of body Cl− stores. Serum [HCO3−] returns to normal when sufficient Cl− is given to replenish losses. Chloride-depletion metabolic alkalosis always causes concomitant K+ depletion through renal K+ losses, but Cl− administration can correct the alkalosis even if the K+ deficit is deliberately maintained.7 Despite this experimental finding, secondary K+ depletion plays a key role in maintaining alkalosis. When K+ depletion is maintained, much larger amounts of Cl− are needed to correct the disorder completely.8 The metabolic alkalosis induced by gastrointestinal Cl− losses can be very severe, with reports of serum [HCO3−] greater than 60 mmol/l.9,10 A less severe Cl−-dependent metabolic alkalosis, without evident H+ loss, is generated by administration of thiazide or loop diuretics and by two genetic abnormalities in Cl− reabsorption, Bartter and Gitelman syndromes, as discussed later. When metabolic alkalosis is induced by Cl− depletion and dietary Cl− intake is restricted, a characteristic sequence of changes in electrolyte excretion occurs (see Fig. 13-2).6 Sodium and HCO3− excretion increase transiently, then decrease rapidly to low levels, accompanied by an abnormal increase in K+ excretion. The increase in K+ excretion is also transient but still induces significant K+ depletion. In the new steady state, urinary K+ excretion matches intake despite depletion of body K+ stores (see Fig. 13-2). As a result, hypokalemia is a cardinal feature of Cl−-depletion alkalosis. Potassium depletion stimulates H+ secretion in the collecting duct (through H+,K+-ATPase; see Fig. 13-1) and HCO3− reabsorption in the ascending limb of the loop of Henle.11 Potassium depletion also downregulates both the Na+-K+-2Cl− cotransporter and the Na+– Cl− cotransporter,11 increasing Na+ delivery to the collecting duct and further stimulating collecting duct H+ secretion (Fig. 13-3). When K+ deficiency is severe, this effect results in measurable Cl− excretion despite Cl− depletion and sustained metabolic alkalosis even when NaCl is administered.12 Finally, K+ depletion stimulates renal NH4+ production, facilitating the acid excretion needed to sustain metabolic alkalosis. Although the Cl−-HCO3− exchanger is activated by metabolic alkalosis, its activation in turn stimulates collecting duct H+ secretion, resulting in reabsorption of all the secreted HCO3− and continued acid excretion.13 In the steady state, acid excretion matches net acid production despite systemic alkalemia, indicating a failure of downregulation of collecting duct ion transport. The contribution of Cl− and K+ depletion to the maintenance of metabolic alkalosis is of little importance in the clinic because treatment is dictated by the particular setting; some patients need NaCl to replenish ECF volume, and most need KCl to treat K+ depletion. Induction of net K+ loss by severe restriction of dietary K+ intake produces a small but significant increase in serum [HCO3−].8 When dietary Cl− intake is concomitantly restricted, however, the resultant alkalosis is four times as great, illustrating the complementary roles of Cl− and K+ in regulating renal HCO3− reabsorption. Depletion of body K+ stores is probably the most important factor in producing and sustaining the rarer forms of metabolic alkalosis induced by mineralocorticoid excess or apparent mineralocorticoid excess (see later discussion). Metabolic alkalosis produced by primary stimulation of collecting duct ion transport accounts for less than 1% of the clinical incidence of this disorder, and by far the most common cause is primary hyperaldosteronism.14 Primary hyperaldosteronism is characterized by persistently high and unregulated aldosterone secretion, which activates both the epithelial sodium channel (ENaC) and H+-ATPase, regardless of body fluid volume and acid-base status (Fig. 13-4; see also Fig. 13-1). As a result, Na+ reabsorption and H+ secretion are increased directly, and K+ secretion is increased secondarily, depleting body K+ stores. The resultant K+ depletion promotes NH4+ production and activates H+,K+-ATPase activity, further facilitating acid excretion. Sodium retention leads to hypertension and also ensures continued Na+ delivery to the collecting duct, sustaining the cycle of increased reabsorption and increased K+ and H+ secretion. As a result of all these events, metabolic alkalosis is sustained despite normal Cl− intake. Not surprisingly, the degree of alkalosis induced by primary hyperaldosteronism is modulated by both Cl− and K+ intake. Much more rarely, metabolic alkalosis is caused by genetic mutations in the regulation and function of ENaC in the collecting duct (see Etiology and Chapter 49). The kidney responds rapidly to excess alkali by increasing HCO3− excretion, and sustained metabolic alkalosis does not occur unless massive amounts are administered in individuals with normal kidney function. When lesser amounts of NaHCO3 are ingested daily, serum [HCO3−] does not increase unless dietary Cl− intake is severely restricted.15 If HCO3− excretion is impaired as a result of kidney failure, even minimal daily alkali administration can cause a sustained metabolic alkalosis independent of Cl− intake.16 End-stage renal disease is the ultimate model of impaired HCO3− excretion, and any added alkali remains in the body until it is consumed by buffering the strong acids produced by protein metabolism (endogenous acid production). Regardless of the cause, blood pH increases in patients with metabolic alkalosis and elicits secondary hypoventilation, increasing Paco2. The response is a potent one, occurring despite the concomitant development of hypoxemia. On average, Paco2 increases by 0.7 mm Hg (0.1 kP) for each 1 mmol/l increase in serum [HCO3−]. Assuming a normal [HCO3−] of 24 mmol/l and Paco2 of 40 mm Hg, the predicted Pco2 for any given serum [HCO3−] in metabolic alkalosis can be calculated as follows:
Metabolic Alkalosis
Definition
Bicarbonate Transport in the Kidney
Pathophysiology of Metabolic Alkalosis
Secondary Stimulation of Collecting Duct Ion Transport
Chloride Depletion
Potassium Depletion
Primary Stimulation of Collecting Duct Ion Transport
Exogenous Alkali
Secondary Response to Alkalemia Induced by Bicarbonate Retention
Metabolic Alkalosis
Chapter 13