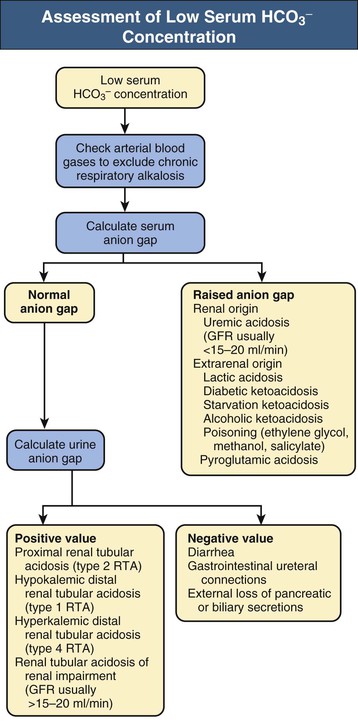
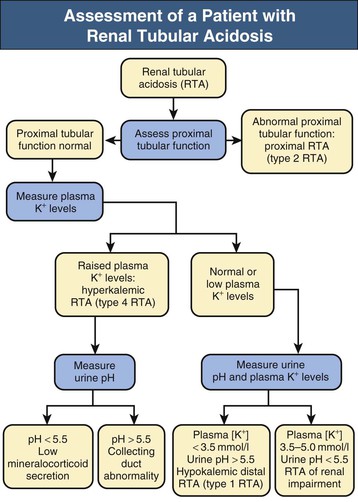
Biff F. Palmer Metabolic acidosis is defined as a low arterial blood pH in conjunction with a reduced serum bicarbonate concentration, [HCO3−]. Respiratory compensation results in a decrease in arterial carbon dioxide tension (Paco2). A low serum [HCO3−] alone is not diagnostic of metabolic acidosis because it also results from the renal compensation to chronic respiratory alkalosis. Measurement of the arterial pH differentiates between these two possibilities. Box 12-1 shows the expected compensatory responses for metabolic and respiratory acid-base disorders.1 After the diagnosis of metabolic acidosis is confirmed, the first step in the examination of the patient is to calculate the serum anion gap. The anion gap equals the difference between the plasma concentrations of the major cation, sodium ([Na+]), and the major measured anions, chloride and bicarbonate ([Cl−] and [HCO3−]), and is given by the following formula: In healthy individuals, the normal value of the anion gap is approximately 12 ± 2 mmol/l. Because many of the unmeasured anions consist of albumin, the normal anion gap is decreased by approximately 4 mmol/l for each 1 g/dl decrease in the serum albumin concentration below normal. The total number of cations must equal the total number of anions, so a decrease in the serum HCO3− concentration must be offset by an increase in the concentration of other anions. If the anion accompanying excess H+ is Cl−, the decrease in serum [HCO3−] is matched by an equal increase in serum [Cl−]. This acidosis is classified as a “normal anion gap” or a “non–anion gap” or a hyperchloremic metabolic acidosis. By contrast, if excess H+ is accompanied by an anion other than Cl−, the decreased [HCO3−] is balanced by an increase in the concentration of the unmeasured anion. [Cl−] remains the same. In this setting, the acidosis is said to be a “high anion gap” or “anion gap” metabolic acidosis. The normal value for the anion gap has tended to fall over time because of changes in how serum Na+ and Cl− are measured.2 Flame photometry for Na+ measurement and a colorimetric assay for Cl− have been replaced by the use of ion-selective electrodes, with which the serum Na+ values have largely remained the same, whereas the serum Cl− values have tended to be higher. As a result, the normal value for the anion gap has decreased to as low as 6 mmol/l in some reports. Recognizing this change, some laboratories have adjusted the calibration set point for Cl− to return the normal value for the anion gap to the 12 ± 2 mmol/l range. The clinician needs to be aware that the average anion gap and range of normal values will vary in different facilities. Figure 12-1 provides a recommended approach to a patient with metabolic acidosis and lists the common causes of metabolic acidosis according to the anion gap. A non–anion gap metabolic acidosis can result from either renal or extrarenal causes. Renal causes of metabolic acidosis occur when renal bicarbonate generation, which results from net acid excretion, does not balance the loss of bicarbonate and other alkali buffers consumed in the buffering of normal endogenous acid production. This failure of net acid excretion is termed renal tubular acidosis (RTA). Extrarenal causes occur when exogenous acid loads, endogenous acid production, or endogenous bicarbonate losses are elevated and exceed renal net acid excretion. The most common extrarenal cause of non–anion gap metabolic acidosis is chronic diarrhea. Renal and extrarenal causes of metabolic acidosis can be distinguished by measuring urinary ammonia excretion.3 The primary response of the kidney to metabolic acidosis is to increase urinary ammonia excretion, each millimole of urinary ammonia excreted resulting in the generation of 1 mmol of “new” bicarbonate. Thus, renal causes of metabolic acidosis are characterized by low urinary ammonia excretion rates. In contrast, in extrarenal metabolic acidosis, urinary ammonia excretion is elevated. Because most laboratories do not measure urinary ammonia, one can indirectly assess ammonia excretion by measuring the urinary anion gap (UAG): The UAG is normally a positive value, ranging from +30 to +50 mmol/l. A negative value for the UAG suggests increased renal excretion of an unmeasured cation (i.e., cation other than Na+ or K+). One such cation is NH4+. With chronic metabolic acidosis due to extrarenal causes, urinary ammonia concentrations, in the form of NH4Cl, can reach 200 to 300 mmol/l. As a result, the measured cation concentration will be less than the measured anion concentration, which includes the increased urinary Cl−, and the UAG will be less than zero and frequently less than −20 mmol/l. The UAG only indirectly reflects the urinary ammonia concentration and, if other unmeasured ions are excreted, can give misleading results. Examples include diabetic ketoacidosis, associated with substantial urinary excretion of sodium keto acid salts, and toluene exposure (discussed later), associated with increased urinary excretion of sodium hippurate and sodium benzoate. In these settings, the UAG value may remain positive despite an appropriate increase in urinary ammonia excretion because of the increased urinary excretion of Na+ acid-anion salts. In most patients, these conditions are associated with an elevated–anion gap metabolic acidosis, not a non–anion gap metabolic acidosis, and thus are easily distinguishable from diarrhea-induced metabolic acidosis. Urine pH, in contrast to the UAG, does not reliably differentiate acidosis of renal origin from that of extrarenal origin. For example, an acid urine pH does not necessarily indicate an appropriate increase in net acid excretion. If renal ammonia metabolism is inhibited, as occurs with chronic hyperkalemia, there is decreased ammonia available in the distal nephron to serve as a buffer, and small amounts of distal H+ secretion can lead to a significant urine acidification. In this setting, the urine pH is acid, but net acid excretion is low because of the low ammonia excretion. Similarly, alkaline urine does not necessarily imply a renal acidification defect. In conditions in which ammonia metabolism is stimulated, distal H+ secretion can be massive and yet the urine remains relatively alkaline because of the buffering effects of ammonia. An overall approach to patient assessment for workup of metabolic acidosis of renal origin is shown in Figure 12-2. Normally, 80% to 90% of the filtered load of HCO3− is reabsorbed in the proximal tubule. In proximal (type 2) RTA, the proximal tubule has a decreased capacity to reabsorb filtered bicarbonate. When serum bicarbonate concentration is normal or nearly normal, the amount of bicarbonate filtered by the glomerulus exceeds proximal tubule bicarbonate reabsorptive capacity. When this happens, there is increased bicarbonate delivery to the loop of Henle and distal nephron that exceeds their capacity to reabsorb bicarbonate. As a result, some filtered bicarbonate appears in the urine. The net effect is that serum [HCO3−] decreases. Eventually, the filtered bicarbonate load decreases to the point at which the proximal tubule is able to reabsorb sufficient filtered bicarbonate that the bicarbonate load to Henle loop and the distal nephron is within their reabsorptive capacity. When this occurs, no further bicarbonate is lost in the urine, net acid excretion normalizes, and a new steady-state serum [HCO3−] develops, although at a lower-than-normal level. Hypokalemia is present in proximal RTA. Renal NaHCO3 losses lead to intravascular volume depletion, which in turn activates the renin-angiotensin-aldosterone system. Distal Na+ delivery is increased as a result of the impaired proximal reabsorption of NaHCO3. Because of the associated hyperaldosteronism and increased distal nephron Na+ reabsorption, there is increased K+ secretion. The net result is renal potassium wasting and the development of hypokalemia. In the steady state, when virtually all the filtered HCO3− is reabsorbed in the proximal and distal nephron, renal potassium wasting is less, and the degree of hypokalemia tends to be mild. Proximal RTA may occur as an isolated defect in acidification, but type 2 typically occurs in the setting of widespread proximal tubule dysfunction (Fanconi syndrome). In addition to decreased HCO3− reabsorption, patients with Fanconi syndrome have impaired reabsorption of glucose, phosphate, uric acid, amino acids, and low-molecular-weight proteins. Various inherited and acquired disorders have been associated with the development of Fanconi syndrome and proximal RTA (Box 12-2). The most common inherited cause in children is cystinosis (see Chapter 50). Most adults with Fanconi syndrome have an acquired condition that is related to an underlying dysproteinemic condition, such as multiple myeloma. Skeletal abnormalities are common in these patients. Osteomalacia can develop from chronic hypophosphatemia caused by renal phosphate wasting if Fanconi syndrome is present. These patients may also have a deficiency in the active form of vitamin D because of an inability to convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D in the proximal tubule. In contrast to distal RTA, proximal RTA is not associated with nephrolithiasis or nephrocalcinosis. One exception is the use of topiramate,4,5 an antiepileptic drug that is increasingly used to treat a variety of neurologic and metabolic disorders. The drug exerts an inhibitory effect on renal carbonic anhydrase activity, resulting in a proximal acidification defect similar to that observed with acetazolamide. Topiramate also is associated with hypocitraturia, hypercalciuria, and elevated urine pH, leading to an increased risk of kidney stone disease. Proximal RTA should be suspected in a patient with a normal anion gap acidosis and hypokalemia who has an intact ability to acidify the urine to below 5.5 while in a steady state.6 Proximal tubular dysfunction, such as euglycemic glycosuria, hypophosphatemia, hypouricemia, and mild proteinuria, helps support this diagnosis. The UAG is greater than zero, indicating the lack of increase in net acid excretion. Treatment of proximal RTA is difficult. Administration of alkali increases serum [HCO3−], which increases urinary bicarbonate losses and thereby minimizes subsequent increases in the serum [HCO3−]. Moreover, the increased distal sodium load, in combination with increased circulating plasma aldosterone, results in increased renal potassium wasting and worsening hypokalemia. As a result, substantial amounts of alkali, often in the form of a potassium salt, such as potassium citrate, are required to prevent worsening hypokalemia. Children with proximal RTA should be aggressively treated to normalize their serum [HCO3−] to minimize growth retardation. These children may require large amounts of alkali therapy, typically 5 to 15 mmol/kg/day. Adults with proximal RTA are frequently not treated as aggressively as children are because of the lack of systemic metabolic abnormalities or bone disease. Many clinicians administer alkali therapy if serum [HCO3−] is less than 18 mmol/l to prevent severe acidosis. Whether more aggressive therapy to normalize serum [HCO3−] is beneficial remains unknown. However, the large amounts of alkali required, about 700 to 1000 mmol/day for a 70-kg individual, make this approach problematic. In contrast to proximal RTA, patients with distal RTA are unable to acidify their urine, either under basal conditions or in response to metabolic acidosis.7,8 Type 1 RTA results from a reduction in net H+ secretion in the distal nephron, which leads to continued urinary bicarbonate losses and prevents urinary acidification, thereby minimizing titratable acid excretion and urinary ammonia excretion. As a result, these patients are unable to match net acid excretion to endogenous acid production, and acid accumulation ensues. The subsequent metabolic acidosis stimulates reabsorption of bone matrix to release the calcium alkali salts present in bone. During prolonged periods, this can result in progressive osteopenia in adults and in osteomalacia in children. Distal RTA can be caused by either impaired H+ secretion (secretory defect) or an abnormally permeable distal tubule, resulting in increased backleak of normally secreted H+ (gradient defect); it may be genetic or acquired. Certain medications, especially amphotericin, result in increased backleak of protons across the apical plasma membrane, leading to a gradient defect form of distal RTA. For patients with a secretory defect, the inability to acidify the urine below pH 5.5 results from abnormalities in any of the proteins involved in collecting duct H+ secretion. Some patients may have an isolated defect in the H+,K+-ATPase that impairs H+ secretion and K+ reabsorption.9 A defect confined to the vacuolar H+-ATPase also results in renal potassium wasting.10 The development of systemic acidosis tends to diminish net proximal fluid reabsorption with an increase in distal delivery, resulting in volume contraction and activation of the renin-aldosterone system (RAS). Increased distal Na+ delivery coupled to increased circulating levels of aldosterone then leads to increased renal K+ secretion. Defects in the basolateral anion exchanger (AE1) can also cause distal RTA. In this case, the lack of basolateral HCO3− exit leads to intracellular alkalinization, which inhibits apical proton secretion. Patients with distal RTA have low ammonia secretion rates. The decreased secretion is caused by the failure to trap ammonia in the tubular lumen of the collecting duct as a result of the inability to lower luminal fluid pH. In addition, there is often impaired medullary transfer of ammonia because of interstitial disease. Interstitial disease is frequently present in such patients through an associated underlying disease or as a result of nephrocalcinosis or hypokalemia-induced interstitial fibrosis. In contrast to proximal RTA, nephrolithiasis and nephrocalcinosis are common.11 Urinary Ca2+ excretion is high secondary to acidosis-induced bone mineral dissolution. Luminal alkalinization also inhibits calcium reabsorption, resulting in further increases in urinary calcium excretion.12 Calcium phosphate solubility is also greatly lowered at alkaline pH, and calcium phosphate stone formation is accelerated. Stone formation is further enhanced as a result of low urinary citrate excretion. Citrate is metabolized to HCO3−, and its renal reabsorption is stimulated by metabolic acidosis, thereby minimizing the severity of metabolic acidosis. Urinary citrate also chelates urinary calcium, decreasing ionized calcium concentrations. Accordingly, the decreased citrate excretion that occurs in chronic metabolic acidosis due to distal RTA further contributes to both nephrolithiasis and nephrocalcinosis. Distal RTA may be a primary disorder, either idiopathic or inherited, but it most often occurs in association with a systemic disease, one of the most common of which is Sjögren syndrome (Box 12-3). Hypergammaglobulinemic states as well as drugs and toxins may also cause this disorder. A common cause of acquired distal RTA is glue sniffing. Inhalation of toluene from the fumes of model glue, spray paint, and paint thinners can give rise to hypokalemic normal gap acidosis through multiple mechanisms. First, toluene inhibits collecting duct proton secretion. Second, metabolism of toluene produces the organic acids hippuric and benzoic acid. These are buffered by sodium bicarbonate, resulting in metabolic acidosis and the production of sodium hippurate and sodium benzoate. If plasma volume is normal, these salts are rapidly excreted in the urine, and a non–anion gap metabolic acidosis develops. If plasma volume is decreased, urinary excretion is limited, these salts accumulate, and an anion gap metabolic acidosis develops. Distal RTA should be considered in all patients with a non–anion gap metabolic acidosis and hypokalemia who have an inability to lower the urine pH maximally. A urine pH above 5.5 in the patient with systemic acidosis suggests distal RTA, and a UAG value greater than zero is confirmatory. Depending on the duration of the distal RTA, the metabolic acidosis can be mild or very severe, with a serum [HCO3−
Metabolic Acidosis
Definition
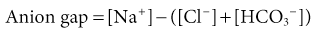
Non–Anion Gap (Normal Anion Gap) Metabolic Acidosis
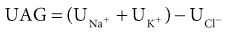
Metabolic Acidosis of Renal Origin
Proximal Renal Tubular Acidosis (Type 2)
Hypokalemic Distal Renal Tubular Acidosis (Type 1)
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Metabolic Acidosis
Chapter 12