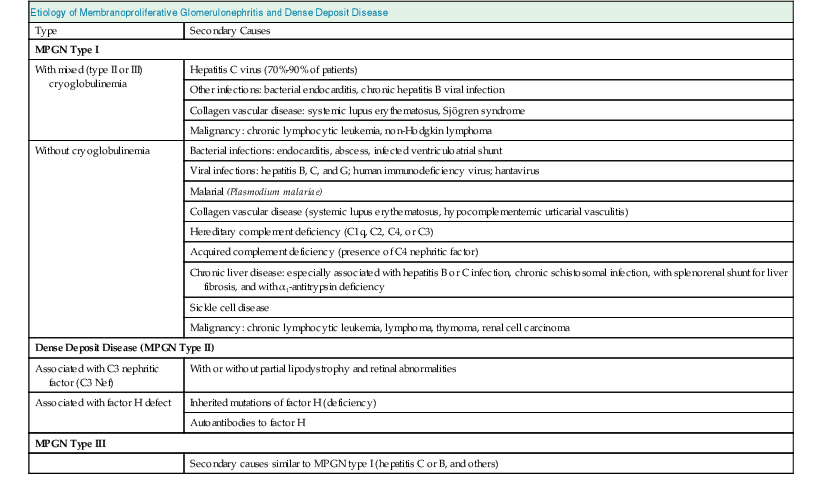
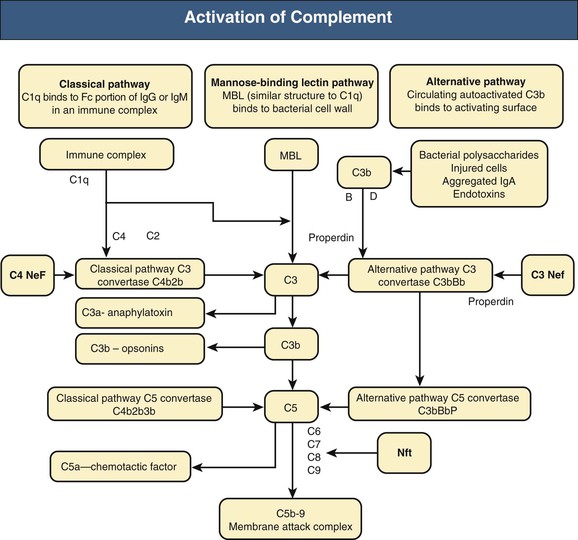
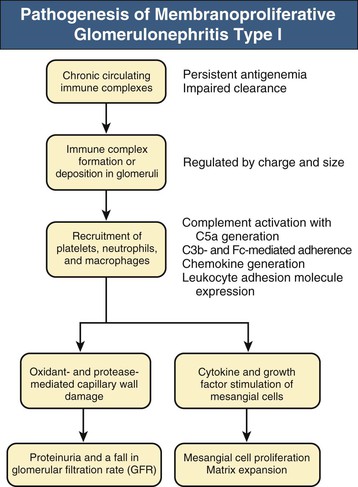
Francesco P. Schena, Charles E. Alpers Membranoproliferative glomerulonephritis (MPGN), or mesangiocapillary glomerulonephritis, is characterized by persistent antigenemia and circulating immune complexes, diffuse proliferative lesions involving both the mesangium and the peripheral capillary walls, and widening of the capillary loops, often with a double-contoured appearance expressing a capillary wall remodeling. MPGN may be idiopathic, but primary MPGN is increasingly recognized as a diagnosis of exclusion, with many previously “idiopathic” cases now found to be secondary to chronic infections, cryoglobulinemia, or systemic autoimmune disorders that result in aberrant immune complex formation. On the basis of electron microscopy (EM) findings, MPGN has been traditionally classified into three categories. Type I was characterized by immune deposits in the subendothelial space and in the mesangium. Type II, now known as dense deposit disease (DDD), was characterized by dense deposits within the mesangium and in the basement membranes. Type III was considered a variant of type I (see Pathology). However, a more modern classification has emerged of MPGN based on underlying pathogenetic mechanisms. In this approach, which we follow in this chapter, MPGN is used to describe an immune complex disorder that includes most of those cases formerly considered MPGN type I and some cases of MPGN type III of Burkholder type. Cases of the former MPGN type II (DDD) and type III of Strife and Anders type are considered part of the spectrum of diseases now called C3 glomerulopathies, discussed separately in Chapter 22. Some cases formerly considered MPGN type I, in which deposits of complement (C3) are detected but not immunoglobulin, are also best considered as belonging to the class of C3 glomerulopathies, although evidence of alternative complement pathway dysregulation may not always be available for these cases.1 Membranoproliferative glomerulonephritis denotes a pattern of injury that is similar, whether the disease is primary or not. Therefore, the histologic diagnosis of MPGN should initiate a search for secondary causes2 (Table 21-1). In older adults (>30 years), MPGN is frequently associated with cryoglobulinemia and hepatitis C virus (HCV) infection. Membranoproliferative GN is most likely to occur in the setting of chronic immune complex diseases, for example, when the host cannot eliminate a foreign antigen effectively despite a humoral response. This may account for the MPGN observed with chronic blood-borne viral (hepatitis C and hepatitis B), bacterial (endocarditis, infected ventriculoatrial shunt), and malarial infections (see Chapter 57). A histologic pattern resembling MPGN can also be observed in chronic immune complex diseases such as lupus. Chronic immune complex disease and MPGN may also occur if the host has a defect in clearing immune complexes, as in complement deficiency, or when the reticuloendothelial system is impaired, as occurs with liver or splenic disease. Hereditary deficiencies of the classical pathway of complement (C1q, C2, C4) and of C3 may predispose to lupus and bacterial infections, then to development of MPGN. The disease is also associated with some malignant neoplasms, especially chronic lymphocytic leukemia (CLL) and lymphoma. Membranoproliferative GN results from the glomerular deposition of immune complexes or from in situ immune complex formation (see Chapter 16). These complexes preferentially localize in the mesangium and subendothelial space of the capillary walls. Once localized, they typically activate complement through the classical pathway, leading to the generation of chemotactic factors (C5a), opsonins (C3b), and the membrane attack complex (C5b-9), with corresponding lowering of circulating C3 and C4 serum levels. In some patients, complement activation may be enhanced by C4 nephritic factors; in other cases, activation of complement may occur by the mannose-binding lectin (MBL) pathway3 (Fig. 21-1). Complement activation results in the release of chemotactic factors that promote leukocyte accumulation and direct recruitment of leukocytes by engagement of Fc receptors present on these cells (Fig. 21-2). These features likely are important in the influx of monocytes, a noteworthy feature of MPGN. Leukocytes release oxidants and proteases, mediating capillary wall damage and proteinuria and a decrease in glomerular filtration rate. Cytokines and growth factors released by both exogenous and endogenous glomerular cells lead to mesangial proliferation and matrix expansion. Cryoglobulins (i.e., immunoglobulins) precipitate in the cold and are categorized as types I to III (Table 21-2). The mixed cryoglobulinemias (types II and III) are most often associated with MPGN and have been strongly associated with chronic HCV infection in up to 80% to 90% of patients with cryoglobulinemic vasculitis.4 The non-HCV cases have been associated with other infections (chronic hepatitis B, bacterial endocarditis), autoimmune diseases (systemic lupus), and other immunologic disorders (notably postinfectious GN). CLL may also be associated with cryoglobulinemia and MPGN, but cases of CLL and lymphoma also have been associated with MPGN in the absence of cryoglobulinemia.5 Many patients with HCV infection and cryoglobulinemia have an IgM-κ monoclonal antibody with rheumatoid factor (RF) activity. Complexes of this IgM RF can be found with anti-HCV and IgG, along with HCV peptides, and HCV RNA can be found in cryoprecipitates from patient serum. It has been postulated that the IgM is produced by dysregulated B cells infected with HCV. Cryoglobulinemia does not develop until many years (often >10) after HCV infection; but by the time chronic active hepatitis or cirrhosis develops, as many as 30% to 40% of patients will have circulating cryoglobulins or other evidence of cryoglobulinemia.6 Most of these patients will not develop renal disease, but in some, possibly those in whom the cryoglobulins have an affinity for fibronectin, the cryoglobulins containing HCV antigens will deposit in glomeruli.7 Specific HCV-related proteins have been detected in glomeruli (Fig. 21-3), tubulointerstitium, and vessels in patients with cryoglobulinemic HCV-positive MPGN,8 although difficulties with the antisera available to detect HCV render such findings controversial. Glomerular HCV deposits have displayed two different patterns: (1) a linear homogeneous deposition along glomerular capillary walls, including endothelial and subendothelial spaces, and (2) a granular appearance on immunofluorescence microscopy with distinct deposits in mesangial and paramesangial areas. IgG, IgM, and C3 deposits have a distribution comparable to those of HCV RNA and core protein deposits, suggesting that in cryoglobulinemic HCV-positive MPGN, deposits consist of HCV-containing immune complexes that may directly contribute to renal damage.9 Further pathogenetic insight will likely be provided by new murine models, including the transgenic thymic stromal lymphopoietin (TSLP) mouse, which develops mixed cryoglobulinemia and MPGN.10 Monocytes/macrophages appear to mediate progression of renal damage in this model.11 In North America and Europe, MPGN constitutes less than 5% of all primary glomerulopathies. MPGN accounts for 5% to 10% of primary renal causes of nephrotic syndrome in children and adults.12 Affecting males and females equally, MPGN in the United States is relatively more common in Caucasians than African Americans. MPGN remains the most common cause of GN in many developing countries, especially in Africa and South America (such as Peru). The renal biopsy registries show a declining incidence and prevalence of MPGN in the last 40 years in industrialized countries, whereas it remains common in developing countries, where the occurrence of infections is high. The disease may be familial in rare cases, and different histologic lesions may occur in family members. Membranoproliferative GN may present as microhematuria and non-nephrotic proteinuria (35%), as nephrotic syndrome with minimally depressed renal function (35%), as a chronically progressive GN (20%), or as a nephritic syndrome with rapidly deteriorating renal function with proteinuria and red blood cell casts (10%). Hypertension is present in 50% to 80% of patients, occasionally so severe that the presentation may be confused with that of malignant hypertension. When presenting in childhood, primarily between ages 8 and 14 years, MPGN is usually idiopathic. Previously reported series have frequently included both immune complex–mediated MPGN and DDD together in characterizing the features of MPGN in children. Asymptomatic Japanese children diagnosed through a urinalysis screening program at school had lower blood pressure, proteinuria, and serum creatinine concentration than individuals diagnosed after presenting with symptoms.13 Thus, early identification of the disease by urinary screening may allow early treatment.
Membranoproliferative Glomerulonephritis and Cryoglobulinemic Glomerulonephritis
Definition
Etiology and Pathogenesis
Epidemiology
Clinical Manifestations
Pediatric Population
Adult Population
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Membranoproliferative Glomerulonephritis and Cryoglobulinemic Glomerulonephritis
Chapter 21